017 Define cirrhosis, classify cirrhosis. Describe aetiopathogenesis, 20 Novem 2 PIMS morphology, clinical findings and complications of Alcoholic ber cirrhosis. L.Q 2017 Describe aetiopathology of cirrhosis of liver. 20 March 2 MS UNIVERS L.Q 2
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Finding Sources
Reading File
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Reading File
Loading Image
Reading File
I now have comprehensive content to write a detailed long-question answer. Let me compile it.
Cirrhosis of the Liver - Comprehensive Long Question Answer
1. DEFINITION
Cirrhosis is the diffuse transformation of the entire liver into regenerative parenchymal nodules surrounded by dense bands of fibrous (collagen-rich) scar tissue, resulting in distortion of hepatic architecture and vascular disruption. It represents the morphologic end point of most chronic liver diseases and is associated with varying degrees of hepatocellular dysfunction.
"Cirrhosis refers to the diffuse transformation of the liver into regenerative parenchymal nodules surrounded by fibrous bands."
- Robbins & Kumar Basic Pathology
2. CLASSIFICATION OF CIRRHOSIS
A. Morphological Classification
| Type | Nodule Size | Common Causes |
|---|---|---|
| Micronodular (Laennec's cirrhosis) | Nodules <3 mm, uniform size | Alcohol, biliary obstruction, hemochromatosis |
| Macronodular (post-necrotic) | Nodules >3 mm, variable size | Viral hepatitis (HBV, HCV), autoimmune |
| Mixed | Both micro- and macronodules | Many causes; micronodular may evolve into macronodular |
B. Aetiological Classification
| Category | Examples |
|---|---|
| Alcohol-associated | Chronic alcohol use disorder |
| Viral | Chronic HBV, HCV infection |
| Metabolic | MASLD (formerly NAFLD), Wilson's disease, haemochromatosis, Alpha-1 antitrypsin deficiency |
| Biliary | Primary biliary cholangitis, primary sclerosing cholangitis, secondary biliary cirrhosis |
| Autoimmune | Autoimmune hepatitis |
| Vascular | Budd-Chiari syndrome, cardiac cirrhosis (right heart failure) |
| Cryptogenic | No identifiable cause (~10-15% of cases) |
The leading causes worldwide are chronic HBV, chronic HCV, MASLD, and alcohol-associated liver disease.
3. AETIOPATHOGENESIS OF CIRRHOSIS (General)
Regardless of cause, the shared pathogenetic pathway involves three key processes:
Step 1 - Repeated/Sustained Hepatocellular Injury
Any chronic stimulus (viral, toxic, metabolic, biliary) causes ongoing hepatocyte death via necrosis or apoptosis.
Step 2 - Stellate Cell Activation and Fibrogenesis
- Damaged hepatocytes and Kupffer cells release TGF-β, TNF-α, IL-1, and platelet-derived growth factor (PDGF).
- These cytokines activate hepatic stellate cells (Ito cells) in the space of Disse.
- Activated stellate cells transform into myofibroblast-like cells and produce large amounts of type I and III collagen, fibronectin, and other extracellular matrix (ECM) components.
- Result: progressive perisinusoidal fibrosis leading to "capillarization of sinusoids" (loss of fenestrations in the sinusoidal endothelium), which impairs exchange between blood and hepatocytes.
Step 3 - Regeneration and Nodule Formation
- Surviving hepatocytes undergo regenerative hyperplasia, forming nodules without the normal lobular architecture.
- These nodules are enclosed by fibrous septa that link portal tracts to central veins (bridging fibrosis), disrupting normal lobular architecture.
Step 4 - Vascular Distortion
- Perivenular fibrosis and fibrous obliteration of terminal hepatic venules (phlebosclerosis)
- Veno-occlusive lesions → sinusoidal hypertension → portal hypertension
- Portosystemic shunting: blood bypasses functioning hepatocytes
4. AETIOPATHOGENESIS OF ALCOHOLIC CIRRHOSIS
4.1 Epidemiology
- Alcohol accounts for 48% of cirrhosis worldwide, causing 1.16 million deaths annually
- Only 10-20% of heavy drinkers develop cirrhosis - other cofactors are important
- Threshold: 80 g/day of ethanol (80-proof liquor); cirrhosis typically takes 10-20 years to develop
4.2 Risk Factors for Progression
| Factor | Mechanism |
|---|---|
| Amount/duration | Dose-dependent hepatotoxicity |
| Sex (female) | Lower threshold; estrogen increases gut permeability to endotoxins → Kupffer cell TLR4 activation |
| Genetics | PNPLA3, MBOAT7, TM6SF2 polymorphisms; ALDH2 variants (common in Asians) |
| Obesity/MASLD | Synergistic hepatotoxicity |
| Viral hepatitis (HBV/HCV) | Additive liver injury |
| Malnutrition | Impaired hepatic repair |
| Smoking | Independent risk factor |
| Gut microbiome dysbiosis | Increased intestinal permeability → endotoxaemia |
4.3 Mechanisms of Alcohol-Induced Hepatocyte Injury
Ethanol Metabolism:
Ethanol → (ADH1) → Acetaldehyde → (ALDH) → Acetate
Ethanol → (CYP2E1, induced by chronic drinking) → Acetaldehyde + ROS
Specific injurious mechanisms:
-
Altered redox state (NADH/NAD+ ratio):
- Ethanol oxidation converts NAD+ to NADH
- High NADH/NAD+ inhibits beta-oxidation of fatty acids and gluconeogenesis
- Promotes fatty acid synthesis → hepatic steatosis
-
Acetaldehyde toxicity:
- Induces lipid peroxidation
- Forms acetaldehyde-protein adducts → cytoskeletal and membrane disruption
- May generate neoantigens triggering immune injury
-
CYP2E1-induced oxidative stress:
- Induction by chronic alcohol consumption increases reactive oxygen species (ROS)
- ROS damage cellular proteins, membranes, and mitochondria
- May promote hepatocyte apoptosis
-
Impaired methionine metabolism:
- Alcohol decreases hepatic glutathione levels → sensitisation to oxidative injury
- Homocysteine production → endoplasmic reticulum (ER) stress
-
Gut-derived endotoxaemia (LPS/PAMPs):
- Alcohol increases gut permeability
- Bacterial lipopolysaccharide (LPS) enters portal circulation
- Activates Kupffer cells via TLR4/CD14
- Triggers NLRP3 inflammasome, releasing IL-1β and caspase-1
- Sustained Kupffer cell activation → chronic hepatic inflammation
-
Mitochondrial dysfunction:
- Impaired beta-oxidation → fat accumulation
- Reduced oxidative phosphorylation → cellular energy deficiency
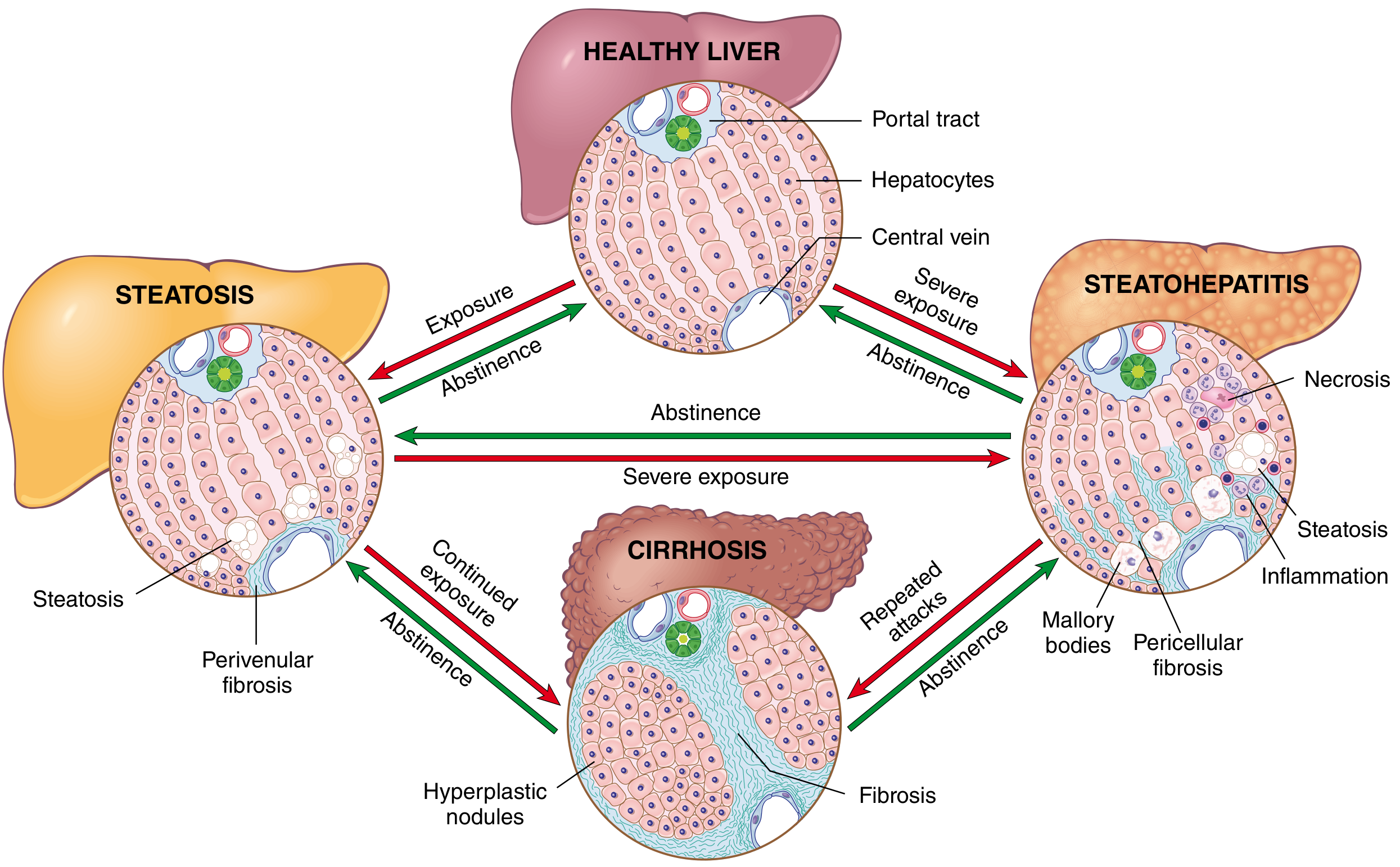
5. MORPHOLOGY OF ALCOHOLIC CIRRHOSIS
5.1 Gross Morphology
Three overlapping, progressive stages:
Stage 1 - Hepatic Steatosis (Fatty Liver):
- Liver enlarged (up to 4-6 kg), soft, yellow, greasy
- Changes begin in centrilobular zone 3 and extend outward
- Reversible with abstinence
Stage 2 - Alcoholic Steatohepatitis:
- Liver may be enlarged or normal in size
- Yellow-tan cut surface; may show irregular nodularity beginning
Stage 3 - Established Alcoholic Cirrhosis:
- Liver is shrunken and firm in end-stage disease
- Surface is diffusely nodular (bumpy texture); normal smooth capsule replaced by irregular surface with depressed scarring and bulging nodules
- Micronodular pattern (Laennec's cirrhosis) initially: nodules <3 mm, uniform in size; with abstinence may evolve into a macronodular or mixed pattern
- Cut surface shows alternating tan-yellow regenerative nodules separated by grey-white fibrous septa
5.2 Microscopic Morphology
Hepatic Steatosis:
- Lipid droplets begin as small (microvesicular) then coalesce into large (macrovesicular) droplets
- Macrovesicular steatosis predominates: fat displaces nucleus to periphery
- Most prominent around central vein (zone 3)
Alcoholic Steatohepatitis (required for diagnosis of hepatitis):
| Feature | Description |
|---|---|
| Ballooned hepatocytes | Swollen hepatocytes with cleared cytoplasm; cytoskeletal damage |
| Mallory-Denk bodies (Mallory hyaline) | Intracytoplasmic eosinophilic inclusions = tangled skeins of ubiquitinated intermediate filaments (keratins 8 and 18) |
| Neutrophilic inflammation | Neutrophils surround ballooned hepatocytes ("satellitosis"); more prominent than in MASLD |
| Lobular lymphocytic infiltrates | Also portal lymphocytic infiltrates |
| Necrosis/apoptosis | Usually spotty; can be confluent in severe cases |
| Pericellular/perisinusoidal fibrosis | "Chicken wire" fibrosis in zone 3; most characteristic early fibrotic pattern |
Established Cirrhosis:
- Fibrous septa interconnect portal tracts and central veins (bridging fibrosis)
- Parenchyma replaced by regenerative nodules with disrupted lobular architecture
- Perivenular fibrosis → phlebosclerosis (fibrous obliteration of terminal hepatic venules)
- Loss of bile ductules in fibrous septa (ductular reaction)
- Variable steatosis and residual inflammation
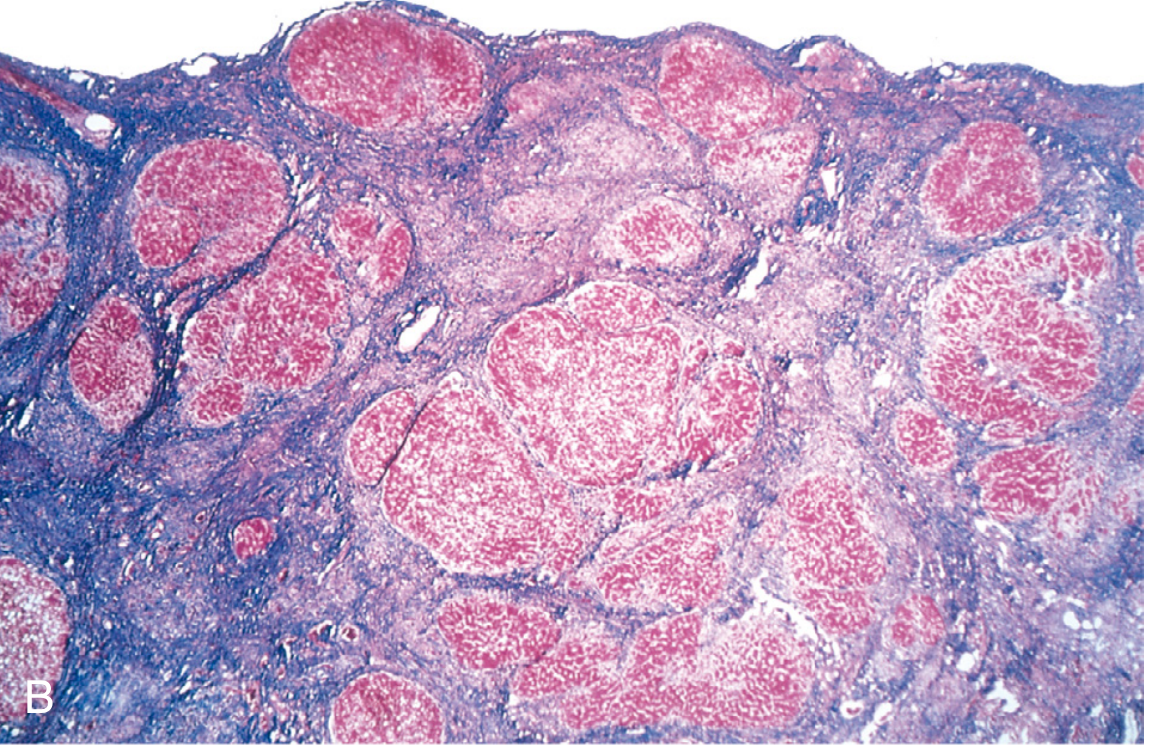
Regression: With prolonged abstinence, fibrous septa become thin and incomplete. Most scars can regress (Fig. 14.6B from Robbins Basic Pathology), but complete regression of cirrhosis is rare.
6. CLINICAL FINDINGS
6.1 Compensated Cirrhosis (~40% asymptomatic)
- Fatigue, weakness, anorexia, weight loss
- Hepatomegaly (early); shrunken liver (late)
- Mild elevation of liver enzymes
6.2 Specific Signs in Alcoholic Cirrhosis
Hepatic findings:
- Hepatomegaly (tender in active hepatitis)
- Jaundice (hyperbilirubinemia)
- Dupuytren's contracture (palmar fibromatosis - classic in alcoholics)
Skin signs (hyperestrogenemia):
- Spider angiomata (arterial spider naevi)
- Palmar erythema
- Gynecomastia, testicular atrophy, loss of libido (males)
- Amenorrhoea (females)
- Rhinophyma (bulbous red nose)
- Jaundice and pruritus (from cholestasis; bile salt accumulation)
Signs of portal hypertension:
- Ascites
- Caput medusae (dilated periumbilical veins)
- Splenomegaly
- Haemorrhoids
Neurological:
- Hepatic encephalopathy (asterixis/flapping tremor, confusion, coma)
- Peripheral neuropathy (from vitamin B1/B12 deficiency)
Other:
- Malnutrition and sarcopenia
- Ecchymoses/bleeding tendency (coagulopathy from decreased clotting factor synthesis)
- Oedema (hypoalbuminaemia)
- Parotid enlargement
6.3 Laboratory Findings
| Test | Finding |
|---|---|
| AST/ALT ratio | >2:1 (rarely >300 IU/L) - characteristic of ALD |
| AST, ALT | Elevated (AST > ALT) |
| GGT | Elevated (sensitive marker) |
| Bilirubin | Elevated |
| Albumin | Decreased |
| PT/INR | Prolonged |
| Platelets | Decreased (hypersplenism) |
| CBC | Anaemia (multifactorial) |
| Serum bilirubin >3 mg/dL | Suggests hepatitis component |
7. COMPLICATIONS
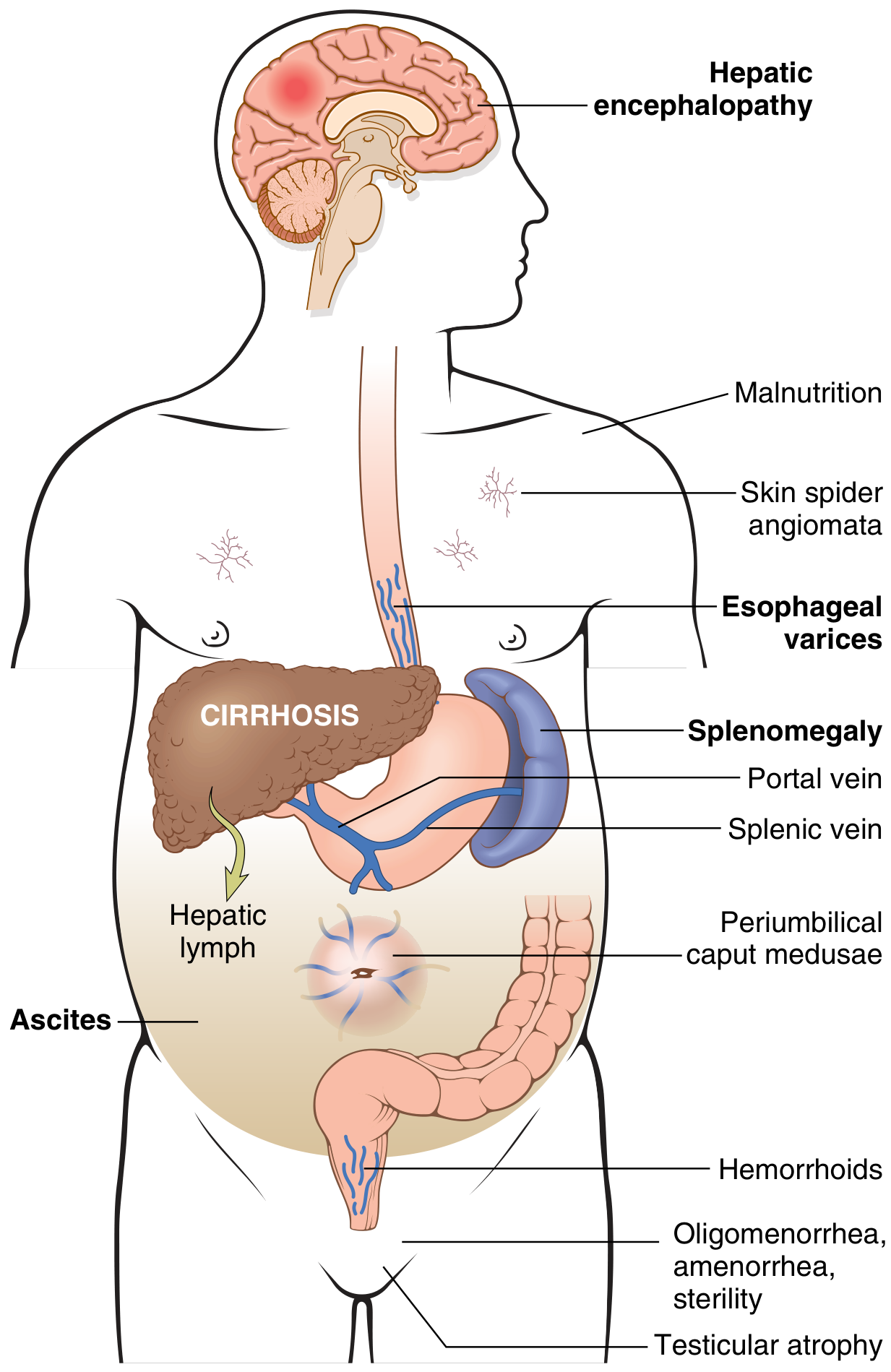
A. Portal Hypertension
- Mechanism: increased sinusoidal resistance (fibrosis + stellate cell contraction) + increased portal blood flow (splanchnic vasodilation)
- Leads to all downstream complications
B. Oesophageal and Gastric Varices
- Develop in ~40% of patients with advanced cirrhosis
- Portosystemic collaterals at oesophago-gastric junction
- Variceal bleeding: massive haematemesis; frequently fatal especially with coagulopathy
C. Ascites
- ~85% of ascites cases are due to portal hypertension from cirrhosis
- Transudate: protein <3 g/dL; serum-to-ascites albumin gradient (SAAG) ≥1.1 g/dL
- Mechanism: portal hypertension + hypoalbuminaemia + renal sodium/water retention (RAAS activation)
- Risk: Spontaneous Bacterial Peritonitis (SBP)
D. Hepatic Encephalopathy
- Accumulation of neurotoxins (ammonia, manganese, false neurotransmitters) due to portosystemic shunting and reduced hepatocyte function
- Clinical stages: from subtle personality changes → confusion → stupor → coma
- Precipitants: GI bleeding, infection, sedatives, hypokalemia, constipation
E. Hepatorenal Syndrome (HRS)
- Functional renal failure without intrinsic renal disease
- Mechanism: liver failure → vasodilator production (NO) → splanchnic vasodilation → decreased renal perfusion → renal vasoconstriction via renin-angiotensin and renal sympathetic activation
- Reversible if liver function restored (e.g., liver transplantation)
F. Splenomegaly and Hypersplenism
- Congestive splenomegaly from portal hypertension
- Spleen may reach 1000 g
- Hypersplenism → thrombocytopenia, anaemia, leukopenia (pancytopenia)
G. Coagulopathy
- Decreased synthesis of clotting factors (I, II, V, VII, IX, X) by failing hepatocytes
- Risk of spontaneous bleeding
H. Hepatocellular Carcinoma (HCC)
- Annual risk: 1-6% in alcoholic cirrhosis
- Cirrhosis of any cause is the single most important risk factor for HCC
- Most chronic liver diseases predispose to HCC
I. Portopulmonary Hypertension
- Pulmonary vasoconstriction due to vasoactive substances bypassing liver
J. Acute-on-Chronic Liver Failure
- Sudden decompensation triggered by: infection, GI bleed, superimposed acute hepatitis, drugs
8. PROGNOSIS
- 5-year survival in abstainers (free of jaundice, ascites, hematemesis): ~90%
- 5-year survival with continued drinking: 50-60%
- Common causes of death:
- Hepatic coma (liver failure)
- Massive GI haemorrhage (variceal bleeding)
- Intercurrent infection (sepsis, SBP)
- Hepatorenal syndrome
- Hepatocellular carcinoma
Child-Pugh score (bilirubin, albumin, PT, ascites, encephalopathy) and MELD score (bilirubin, creatinine, INR) are used to assess severity and guide transplantation eligibility.
Sources: Robbins, Cotran & Kumar - Pathologic Basis of Disease (9780443264528); Robbins & Kumar - Basic Pathology (9780323790185); Harrison's Principles of Internal Medicine 22E (9781265977061)
Discuss the pathology of regenerative and neoplastic helatocellular nodules
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Reading File
Reading File
Finding Sources
Loading Image
Loading Image
I now have comprehensive content from the textbooks. Let me compile the full answer.
Pathology of Regenerative and Neoplastic Hepatocellular Nodules
Hepatocellular nodules form a spectrum from benign regenerative responses through premalignant dysplasia to frank carcinoma. Understanding this spectrum is fundamental to liver pathology.
OVERVIEW: THE NODULAR SPECTRUM
Cirrhotic/Regenerative Nodule
↓
Large Cell Change
Small Cell Change
↓
Low-Grade Dysplastic Nodule
↓
High-Grade Dysplastic Nodule (+ "nodule-in-nodule")
↓
Early / Well-differentiated HCC
↓
Overt / Advanced HCC
Also considered separately as non-cirrhotic regenerative lesions:
- Focal Nodular Hyperplasia (FNH)
- Hepatocellular Adenoma
I. REGENERATIVE NODULES
A. Cirrhotic (Regenerative) Nodules
These are the basic structural units of cirrhosis and are not premalignant per se.
Pathogenesis: Following repeated cycles of hepatocyte injury and death, surviving hepatocytes undergo compensatory regenerative hyperplasia, forming clonal clusters surrounded by fibrous septa.
Morphology:
- Microscopically: nodules composed of hepatocytes with preserved lobular architecture (though simplified); cells are normal in size and cytology
- Surrounded by fibrous bands (portal-to-portal or portal-to-central bridging fibrosis)
- No cytologic atypia
- Micronodular (<3 mm, seen in alcoholic/metabolic disease) or macronodular (>3 mm, seen in post-viral disease)
B. Large Cell Change (Large Cell Dysplasia)
An early at-risk alteration seen in chronic liver disease, particularly viral hepatitis.
Morphology:
- Hepatocytes that are enlarged with large, often atypical nuclei (nuclear enlargement proportional to cytoplasmic enlargement)
- Scattered throughout the lobule; do not form discrete nodules
- Not a truly dysplastic lesion in a strict sense - may represent a senescent, stress-induced change
- Associated with HCV infection and cirrhosis
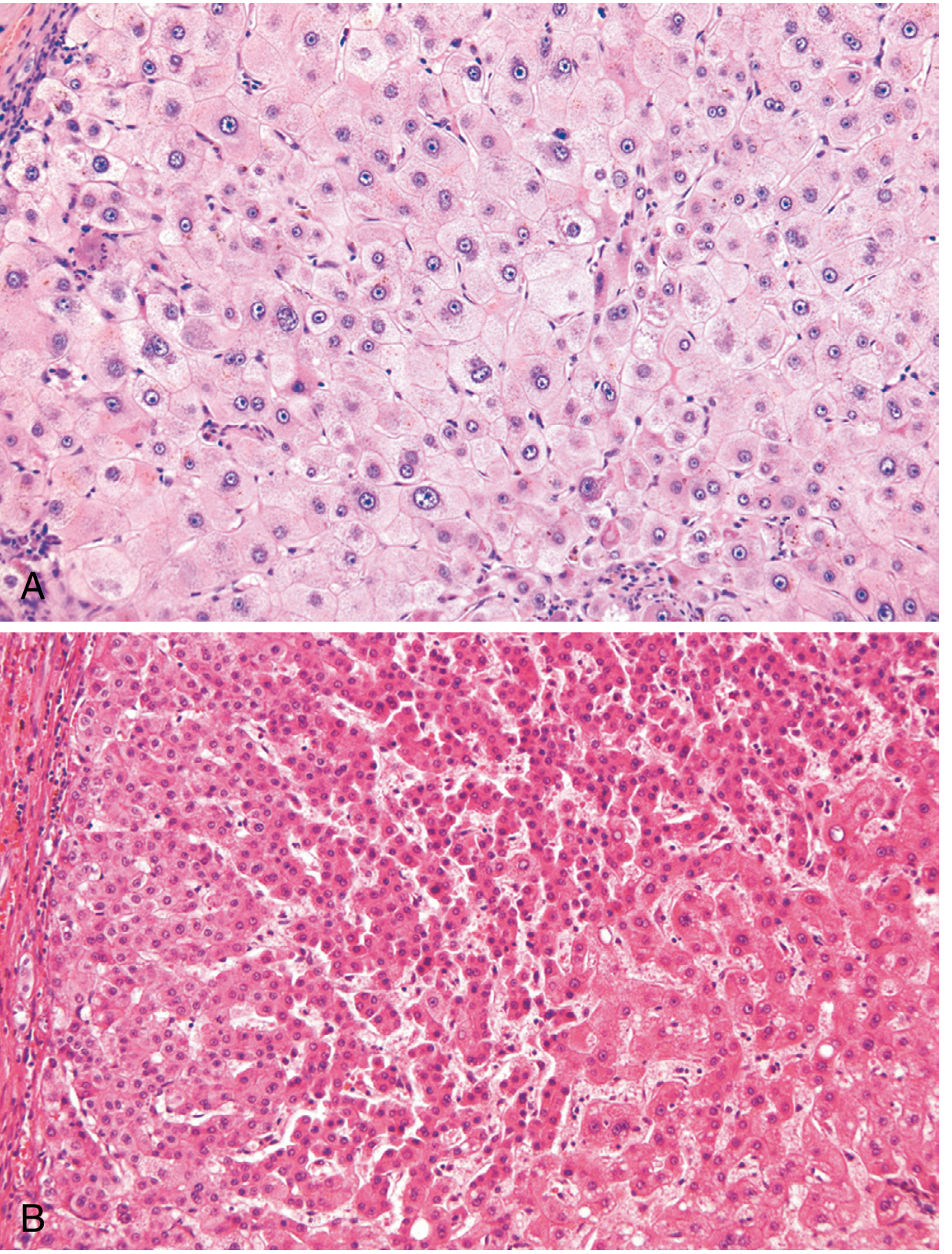
C. Small Cell Change
Considered to carry a higher premalignant risk than large cell change.
Morphology:
- Hepatocytes are smaller than normal with a high nuclear-to-cytoplasmic (N:C) ratio
- Cells grow in thickened plates (2-3 cell plates thick vs normal 1-2)
- Clusters may form and are clonal
- Most closely associated with early HCC and high-grade dysplastic nodules
D. Focal Nodular Hyperplasia (FNH)
A non-neoplastic, regenerative mass lesion occurring in otherwise normal liver. It is the second most common benign hepatic lesion after haemangioma.
Pathogenesis: Thought to arise in response to a pre-existing arterial malformation/vascular anomaly causing local hepatocyte hyperplasia. Not associated with oral contraceptive use (unlike adenoma).
Demographics: Most common in young to middle-aged women; usually asymptomatic, discovered incidentally.
Gross Morphology:
- Well-demarcated but poorly encapsulated nodule, may be several centimeters in diameter
- Central stellate (star-shaped) fibrous scar - pathognomonic feature - from which fibrous septa radiate to periphery
- Normal background liver
Microscopic Morphology:
- Central scar contains large anomalous (thick-walled) arteries and ductular reactions along the fibrous septa
- Hepatocytes appear normal histologically
- No encapsulation; no portal tracts in nodular parenchyma
- No cytologic atypia, no malignant potential
Clinical significance: Benign; rarely causes symptoms; does not require resection unless symptomatic.
II. HEPATOCELLULAR ADENOMA (Benign Neoplasm)
A true benign neoplasm of hepatocytes arising in a non-cirrhotic liver.
Associations:
- Reproductive-age women (historically linked to oral contraceptive pills, now more linked to obesity and metabolic syndrome)
- Anabolic steroid use in men
- Glycogen storage disease
Molecular subtypes:
| Subtype | Mutation | Features |
|---|---|---|
| HNF1α-inactivated | HNF1α loss | Marked steatosis; LFABP absent on IHC; low malignant risk |
| β-catenin-activated | CTNNB1 gain-of-function | High risk of malignant transformation to HCC; nuclear β-catenin on IHC |
| Inflammatory | IL6ST/STAT3 pathway | Marked sinusoidal dilation; serum amyloid A/CRP positive on IHC |
| Unclassified | Unknown | ~10% of cases |
Gross Morphology:
- Usually solitary, well-demarcated mass; no fibrous capsule (or thin capsule)
- May have areas of haemorrhage or necrosis (particularly when large, >5 cm)
- No central scar (distinguishes from FNH)
- Cut surface: pale-tan to yellow
Microscopic Morphology:
- Sheets and cords of hepatocytes resembling normal hepatocytes
- Arterial vascular supply (unpaired arteries without portal tracts) - key diagnostic feature
- No portal tracts within the tumour
- No bile ducts within the tumour
- Variable degree of cytologic atypia depending on subtype; β-catenin-activated type shows most atypia
Complications:
- Haemorrhage and rupture (especially >5 cm) → life-threatening intra-abdominal bleeding
- Malignant transformation to HCC (especially β-catenin-activated subtype)
Management: Resection recommended for male patients (regardless of size), β-catenin-activated tumours, and tumours ≥5 cm.
III. DYSPLASTIC NODULES (Premalignant)
Dysplastic nodules arise in cirrhotic liver and represent clonal proliferations with molecular alterations overlapping with HCC. They are recognised by the International Consensus Group for Hepatocellular Neoplasia.
A. Low-Grade Dysplastic Nodule (LGDN)
- Slightly larger than surrounding cirrhotic nodules
- Mild cytologic atypia: slightly increased N:C ratio, mild nuclear irregularity
- Architecture largely preserved; no increased cell density
- Clonal, but few molecular aberrations
- No increased arterialization
- Low but not zero risk of progression
B. High-Grade Dysplastic Nodule (HGDN)
- Distinctly larger than surrounding cirrhotic nodules
- Significant cytologic atypia: prominent nuclear irregularity, increased N:C ratio, mitotic figures
- Increased cell density; thickened plates
- Small cell change prominent
- Clonal aberrations associated with overt HCC (TERT promoter mutations, etc.)
- Increased unpaired arteries (arterial neovascularisation begins)
- "Nodule-in-nodule" appearance: small foci of overt HCC visible within the dysplastic nodule
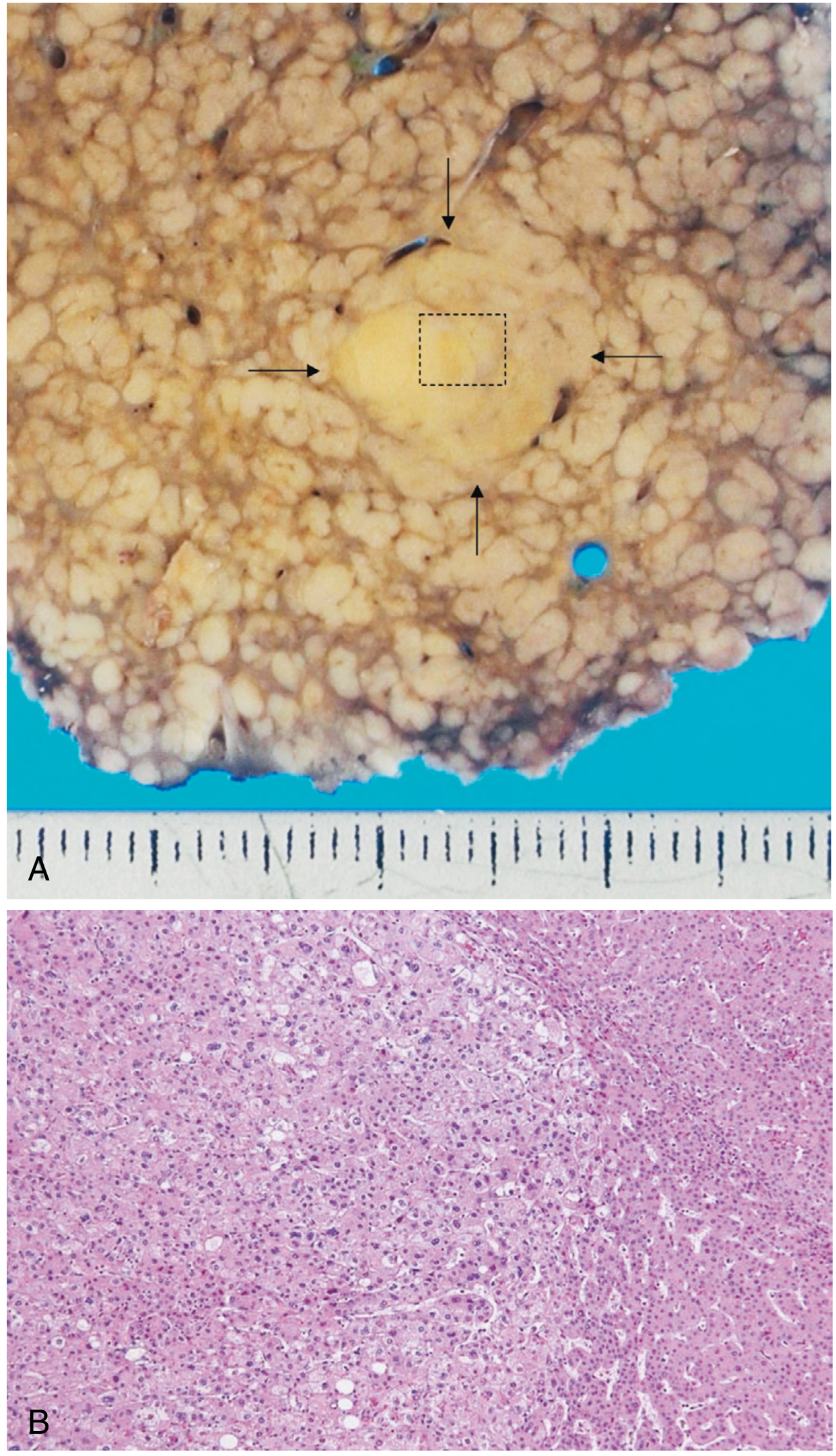
IV. HEPATOCELLULAR CARCINOMA (HCC)
The most common primary malignant liver tumour; represents the neoplastic end of the hepatocellular nodule spectrum.
Epidemiology
- ~5-5.4% of all cancers worldwide
-
85% of cases occur in Asia (SE China, Korea, Taiwan) and sub-Saharan Africa (endemic HBV)
- Male predominance: 3:1 (low-incidence areas) to 8:1 (high-incidence areas)
- Rising in Western countries due to HCV and MASLD
Risk Factors / Aetiology
| Factor | Mechanism |
|---|---|
| HBV (most important globally) | Integration into host genome disrupts tumour suppressors; HBx protein inhibits p53 |
| HCV | Chronic inflammation + cirrhosis drives mutagenesis |
| Aflatoxin B1 (Aspergillus) | Mutagen: G:C→T:A transversion in codon 249 of TP53; synergises with HBV |
| Alcohol | Via cirrhosis; synergises with HBV, HCV |
| MASLD/NAFLD | Via metabolic syndrome and cirrhosis |
| Hereditary haemochromatosis | Iron-induced oxidative DNA damage |
| α1-antitrypsin deficiency | Protein accumulation drives hepatocyte injury |
| Wilson disease | Copper-induced oxidative damage |
| Cirrhosis (any cause) | Chronic regeneration increases mutation acquisition |
Pathogenesis / Molecular Biology
- Chronic liver injury → hepatocyte regeneration → acquisition of driver mutations
- Viruses and toxins are not directly oncogenic; rather, inflammation drives proliferation and mutagenesis
- Key driver mutations in HCC:
- TERT promoter mutations (50-60% of tumours) - upregulate telomerase, prevent replicative senescence
- β-catenin (CTNNB1) activating mutations (~40%) - activate Wnt signalling, promote proliferation
- TP53 inactivating mutations (up to 60%) - loss of cell cycle arrest/apoptosis
- CDKN2A (p16) loss, PI3K/AKT/mTOR pathway activation
- In noncirrhotic liver, HCC can arise from malignant transformation of hepatocellular adenoma (especially β-catenin-activated)
- Aflatoxin causes a characteristic TP53 "hotspot" mutation at codon 249 (Arg→Ser)
Gross Morphology
Three patterns:
- Unifocal (usually large) mass - most common; may replace an entire lobe
- Multifocal, widely distributed nodules of variable size
- Diffusely infiltrative - permeates widely through liver, sometimes involving entire liver; may be mistaken for cirrhosis grossly
Additional features:
- Pale yellow (fatty change) or green (bile production) cut surface
- Tumours >2 cm more likely to show vascular invasion and intrahepatic metastases
- Portal vein invasion: snake-like tumour thrombus extending into portal vein → portal hypertension
- Extension into inferior vena cava and even right ventricle in advanced cases
- Satellite nodules around primary mass (intrahepatic metastases)
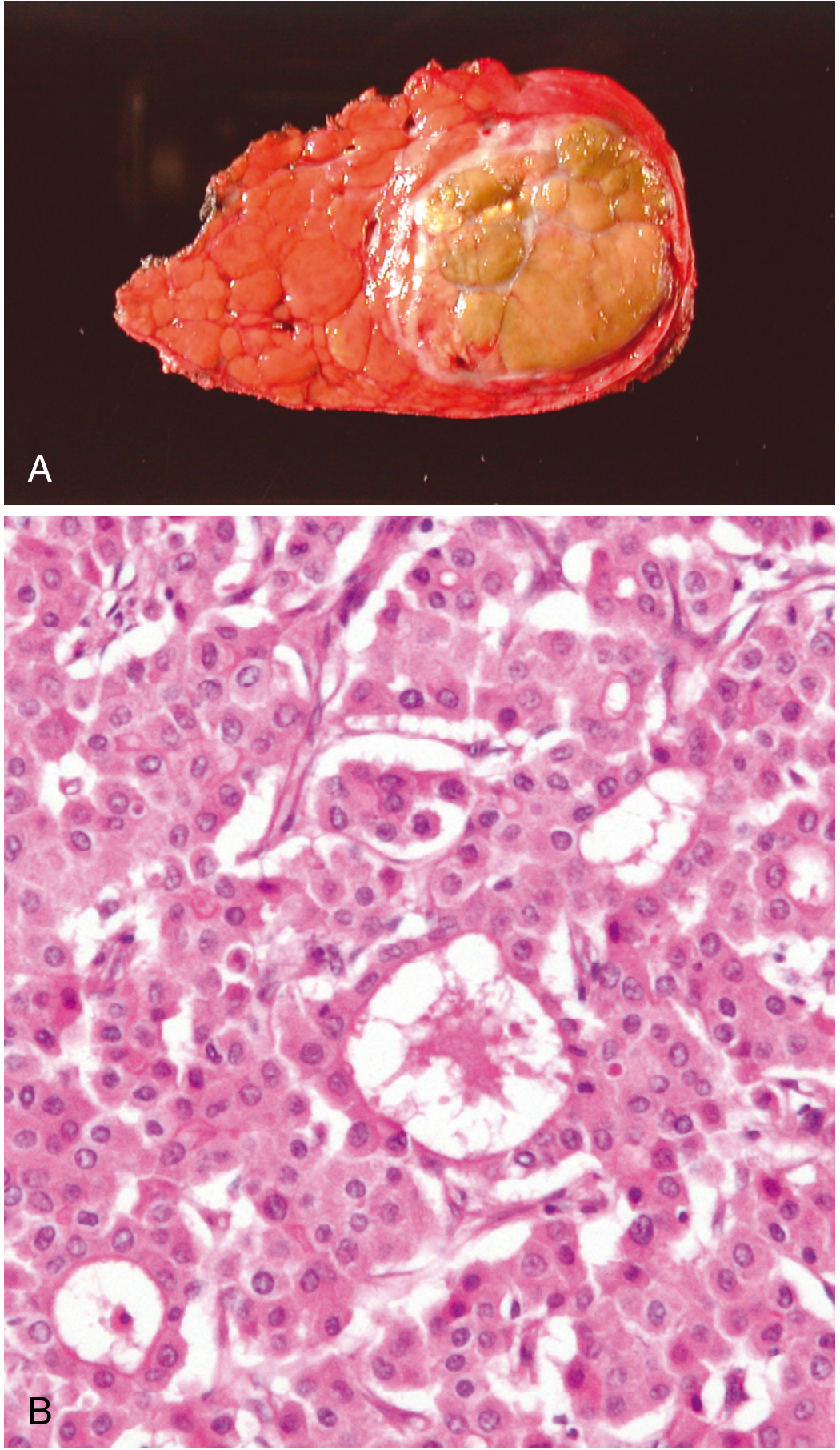
Microscopic Morphology
Histological grades:
| Grade | Features |
|---|---|
| Well-differentiated | Cells closely resemble normal hepatocytes; grow as thick trabeculae (2-3 cell plates) or pseudoglandular/acinar patterns; bile plugs in pseudoacini; mild nuclear atypia |
| Moderately differentiated | Recognisable hepatocytes; more nuclear atypia; mitoses; vascular invasion may be seen |
| Poorly differentiated / Anaplastic | Marked nuclear pleomorphism; giant cells; frequent mitoses; may lose hepatocellular differentiation; spindle cell areas |
Histological growth patterns:
- Trabecular (sinusoidal) - most common; thickened liver cell plates separated by sinusoids lined by flattened endothelial cells
- Pseudoglandular (acinar) - dilated bile canaliculi form pseudoacini; bile plugs present
- Solid/compact - sheets of cells with minimal sinusoidal stroma
- Scirrhous - prominent fibrous stroma (must be distinguished from cholangiocarcinoma)
Immunohistochemistry:
- HepPar-1 (hepatocyte paraffin antigen 1) - most specific hepatocellular marker
- Glypican-3 (GPC3) - sensitive for HCC, especially well-differentiated
- AFP - in tumour cells (correlates with serum levels)
- Arginase-1 - highly sensitive and specific
- Polyclonal CEA - canalicular/bile canalicular pattern (pCEA)
The Fibrolamellar Variant
A distinctive subtype with entirely different clinicopathological features:
- Affects adolescents and young adults (median age 25 years)
- No preexisting liver disease or cirrhosis
- No elevated AFP
- Better prognosis than conventional HCC (up to 40% survive 10 years)
- Molecular hallmark: DNAJB1::PRKACA fusion gene → excessive protein kinase A activity
Morphology (characteristic triad):
- Large polygonal cells with abundant granular eosinophilic cytoplasm (due to abundant mitochondria - oncocytic change)
- Vesicular nuclei with a single prominent nucleolus
- Parallel lamellae of dense collagen (lamellar fibrosis) separating tumour cells
Gross: Large, well-demarcated nodule, often with a central scar (may mimic FNH)
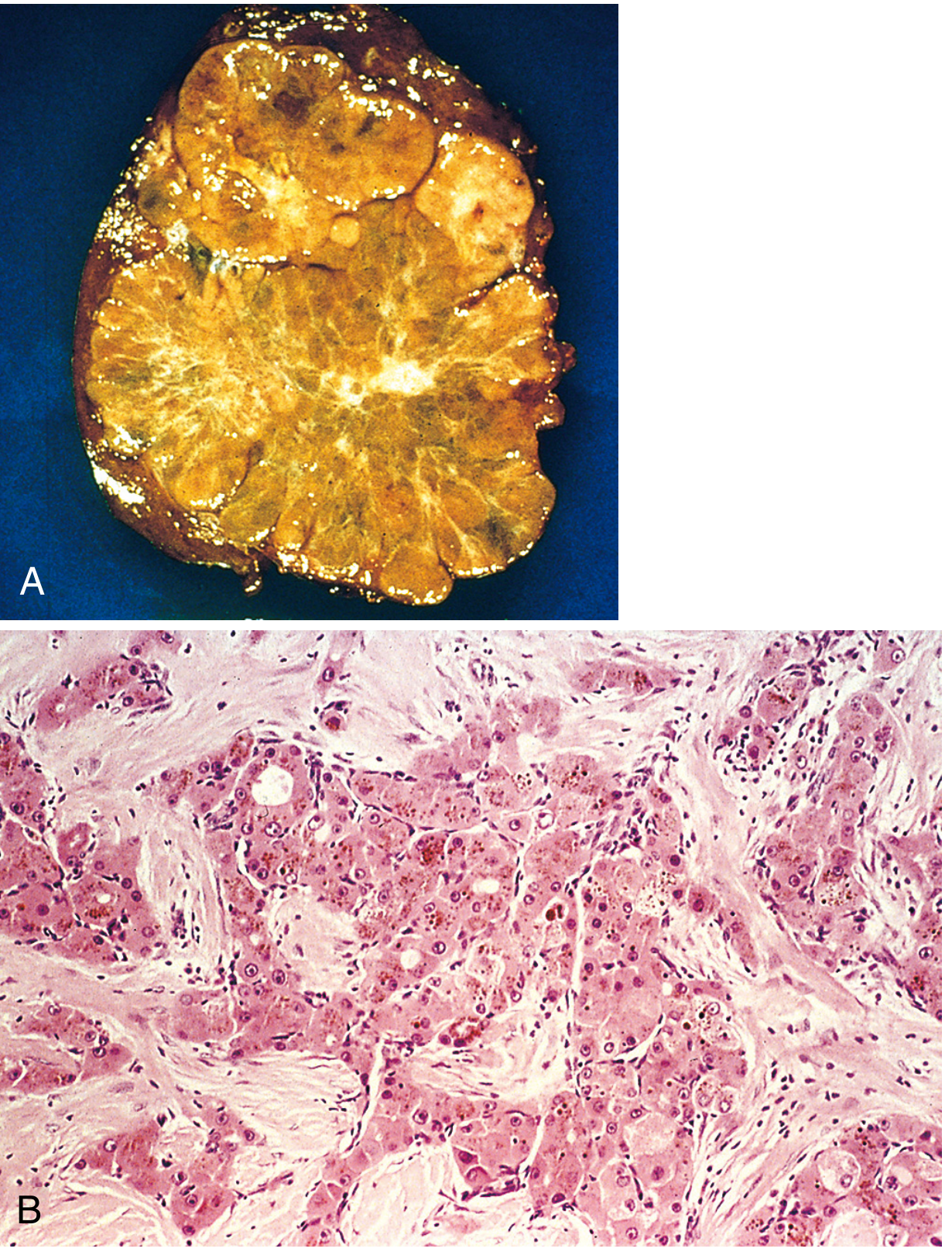
V. COMPARATIVE SUMMARY TABLE
| Feature | Regenerative Nodule (Cirrhosis) | FNH | HCA | Low-Grade DN | High-Grade DN | HCC |
|---|---|---|---|---|---|---|
| Background liver | Cirrhotic | Normal | Normal/non-cirrhotic | Cirrhotic | Cirrhotic | Cirrhotic (85%) |
| Fibrous scar | Surrounding septa | Central stellate scar | No | No | No | No |
| Portal tracts | Present | Absent in nodule | Absent | Reduced | Absent | Absent |
| Bile ducts | Present | Present (ductular rx) | Absent | Reduced | Absent | Absent |
| Arteries | Normal (paired) | Thick-walled central | Unpaired arteries | Slightly increased | Increased unpaired | Markedly increased |
| Cytologic atypia | None | None | Minimal-moderate | Mild | Moderate-marked | Marked |
| Cell plates | 1-2 cells thick | 1-2 | 1-2 | 1-2 | 2-3 | >3 (trabecular) |
| AFP | Normal | Normal | Normal | Normal | Normal/slightly elevated | Elevated (50-60%) |
| Malignant potential | None | None | Low-moderate (β-cat type) | Low | High | N/A (is malignant) |
VI. CLINICAL FEATURES OF HCC
- Symptoms: Upper abdominal pain, malaise, fatigue, weight loss, awareness of abdominal mass
- Jaundice and ascites: Due to underlying cirrhosis or portal vein invasion
- Haemoperitoneum: From spontaneous rupture (especially in sub-Saharan Africa)
- Paraneoplastic syndromes: Hypoglycaemia (insulin-like activity), erythrocytosis (ectopic EPO), hypercalcaemia, hypercholesterolaemia
Laboratory Findings
- Serum AFP elevated in ~50% of advanced HCC; not sensitive for early disease; not elevated in fibrolamellar variant
- AST, ALT, ALP: May be elevated or normal (HCC can arise in relatively preserved liver)
- LDH: Often elevated
Imaging Hallmark
- Contrast-enhanced CT/MRI: Arterial phase enhancement followed by rapid venous washout - pathognomonic of HCC (reflects high arterialization)
- LI-RADS scoring system used for standardised radiological reporting
Prognosis and Treatment
| Stage/Treatment | 5-year Survival |
|---|---|
| Confined to liver (small, resectable) | ~30-40% |
| Extrahepatic spread | ~5% |
| Fibrolamellar variant | Up to 40% at 10 years |
Treatment options:
- Surgical resection (curative; requires adequate liver reserve)
- Liver transplantation (Milan criteria: single tumour ≤5 cm or ≤3 tumours each ≤3 cm; no vascular invasion)
- Radiofrequency ablation / microwave ablation / ethanol injection (unresectable small tumours)
- Transarterial chemoembolisation (TACE) - exploits HCC arterialization
- Sorafenib (kinase inhibitor) - palliative; prolongs survival in advanced disease
- Immune checkpoint inhibitors (atezolizumab + bevacizumab) - current first-line systemic therapy
Causes of death:
- Cachexia
- GI/variceal haemorrhage
- Liver failure / hepatic coma
- Tumour rupture with haemoperitoneum (less common)
Sources: Robbins, Cotran & Kumar - Pathologic Basis of Disease (9780443264528, pp. 807-808); Robbins & Kumar Basic Pathology (9780323790185, pp. 625-630)
Please give pff
Reading File
Running Command
Running Command
Writing File
Running Command
Running Command
Your PDF is ready. Click to download:
Hepatocellular Nodules Pathology
PDF Document · PDF
What's included (4 pages, A4, colour-coded):
- Nodular spectrum overview table - all stages from regenerative nodule → HCC
- Regenerative nodules - cirrhotic nodules, large cell change, small cell change
- FNH - pathogenesis, gross/micro morphology, distinguishing features
- Hepatocellular Adenoma - 4 molecular subtypes with risk stratification table
- Dysplastic nodules - LGDN vs HGDN comparison table; nodule-in-nodule concept
- HCC - epidemiology, risk factors, molecular biology, gross/micro morphology, growth patterns, IHC markers
- Fibrolamellar variant - full characterisation table
- Clinical features, lab findings, imaging hallmark, prognosis, treatment tables
- Master comparative table spanning all nodule types side-by-side
This is a shared conversation. Sign in to Orris to start your own chat.