according to standard literature and latest studies and reporting what is the pathogenesis of PXE
pseudoxanthoma elasticum pathogenesis ABCC6
pseudoxanthoma elasticum PXE pathogenesis 2024 2025 ABCC6 ectopic mineralization mechanism
PMID: 38815804
PMID: 38392293
https://pmc.ncbi.nlm.nih.gov/articles/PMC12004504/
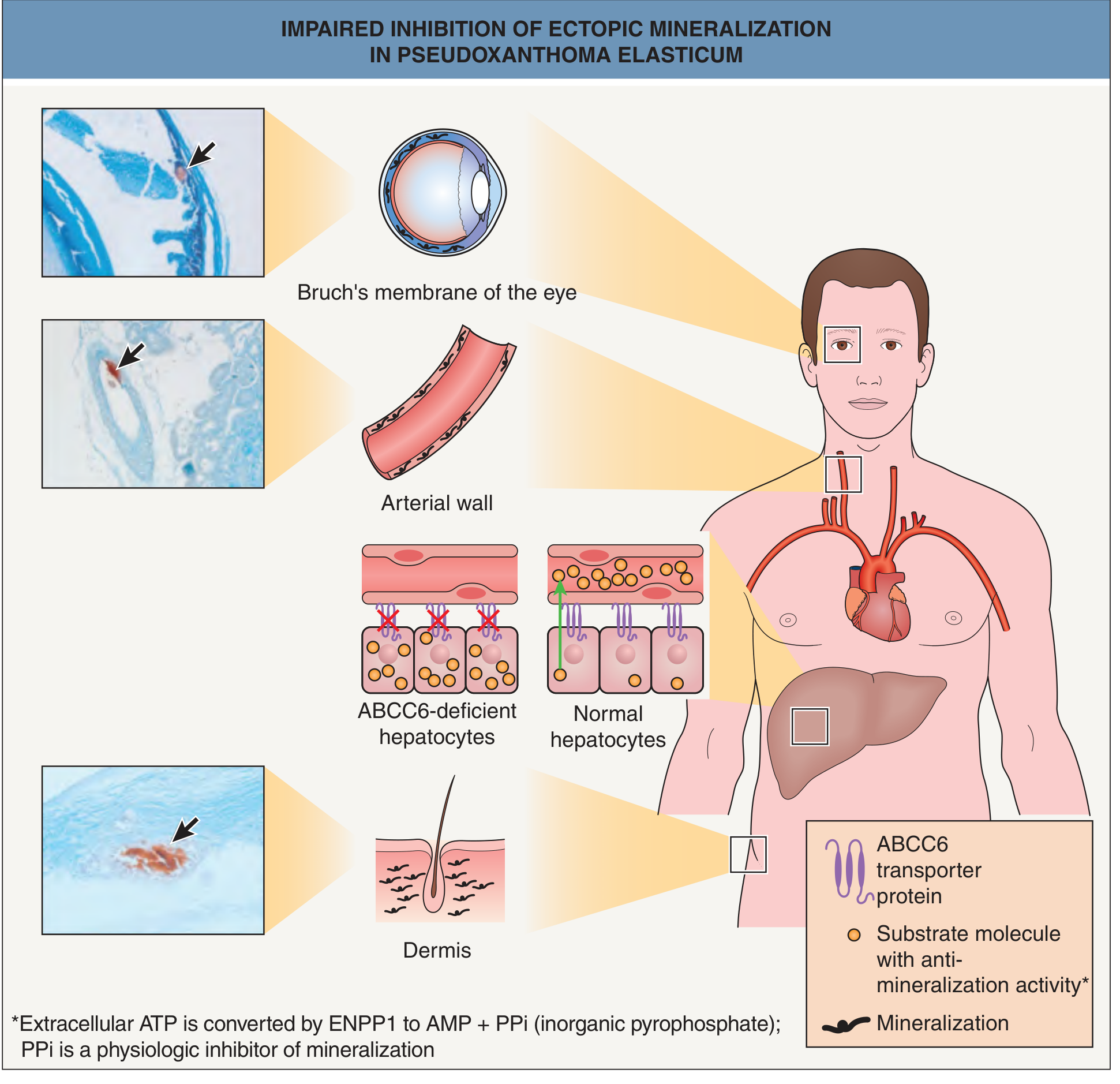
Pathogenesis of Pseudoxanthoma Elasticum (PXE)
1. Genetic Basis: ABCC6 Mutations
2. The Central Metabolic Cascade: ABCC6 → ATP → PPi
Step 1 — Hepatic ATP efflux
Step 2 — ENPP1-mediated hydrolysis
Step 3 — CD73 further metabolizes AMP → adenosine
Step 4 — PPi deficiency leads to unchecked mineralization
3. The Purinergic Disease Concept (2024 Update)
| Disease | Gene | Pathway step affected |
|---|---|---|
| PXE | ABCC6 | ATP efflux from liver |
| GACI (Generalized Arterial Calcification of Infancy) | ENPP1 | ATP → PPi conversion |
| CALJA (Calcification of Joints and Arteries) | NT5E/CD73 | AMP → adenosine |
4. Role of Other Mineralization Inhibitors
Matrix Gla Protein (MGP)
Clinical implication: Vitamin K antagonists should be avoided in PXE patients.
Fetuin-A
Tissue-nonspecific alkaline phosphatase (TNAP)
5. Role of the Bone Marrow and Immune System (Emerging Evidence, 2024)
- The liver is not the sole source of circulating PPi-enabling activity
- The adaptive immune system and local inflammation (lymphangiogenesis was found in PXE skin) contribute substantially to the calcification process
- This opens a new mechanistic dimension beyond the "pure hepatic metabolic disease" model
6. Tissue-Level Pathology: Elastic Fiber Mineralization
- Dermis (mid-portion) → yellow papules, "plucked chicken skin"
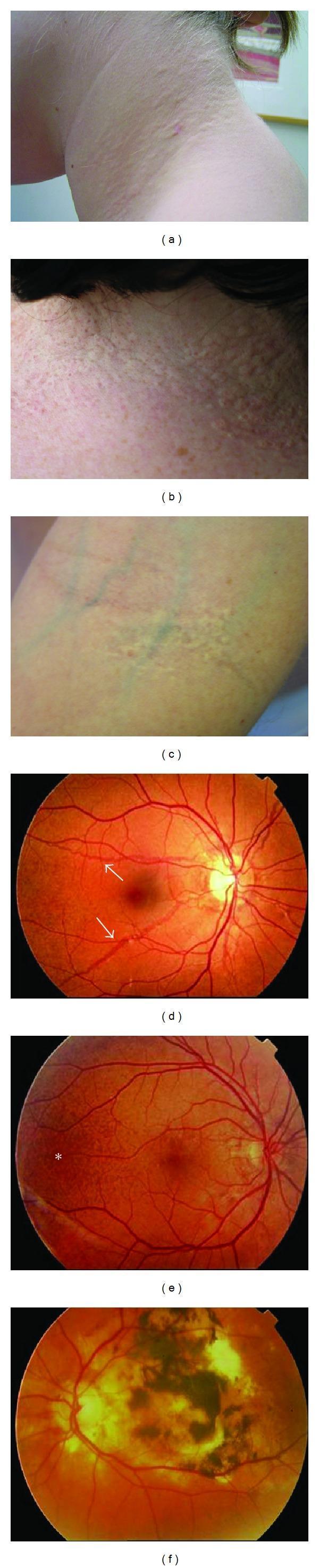
- Bruch's membrane (between RPE and choroid) → angioid streaks, peau d'orange, macular neovascularization
- Medial layer of midsized arteries → premature atherosclerosis, peripheral artery disease, renal hypertension
7. PXE-Like Phenocopies
- β-thalassemia / sickle cell disease: Secondary hemolysis promotes ABCC6 downregulation through iron overload and oxidative stress; PXE-like skin, vascular, and ocular lesions develop
- GGCX mutations: Loss of γ-glutamyl carboxylase → impaired MGP activation → PXE-like phenotype + coagulopathy
- Penicillamine use (Wilson disease): Drug-induced elastin cross-linking disruption → clinicohistologic PXE mimicry
Summary Schematic
ABCC6 mutations (liver)
↓
↓ Hepatic ATP efflux
↓
ENPP1 cannot generate adequate PPi
↓
↓ Plasma PPi + ↓ Carboxylated MGP + ↓ Fetuin-A
↓
Hydroxyapatite nucleation on elastic fibers
↓
Skin (dermis) → yellow papules, skin laxity
Eye (Bruch's membrane) → angioid streaks, CNV, blindness
Arteries (media) → premature atherosclerosis, hypertension, claudication
Key Recent Literature
| PMID | Study | Key Finding |
|---|---|---|
| 38815804 | Pfau et al., Prog Retin Eye Res 2024 | Comprehensive genetics–pathophysiology–clinical review; ocular focus |
| 38392293 | Kauffenstein et al., Biology 2024 | PXE as a "purinergic disease"; ABCC6→ENPP1→CD73→TNAP axis |
| 41519013 | Martin et al., Ann Dermatol Venereol 2026 | REACT-PXE consensus on diagnosis and future research |
| PMC11260544 | Bone marrow transplant study 2024 | Bone marrow ABCC6 contributes to PPi; adaptive immune system involvement |
Textbook sources: Andrews' Diseases of the Skin (9780323547536); Dermatology 2-Volume Set 5e (9780702082252); Fuster & Hurst's The Heart 15e (9781264257560); Emery's Elements of Medical Genetics and Genomics (9780702079665)
what is the metabolic hypothesis of PXE
pseudoxanthoma elasticum metabolic disease hypothesis liver pyrophosphate
PXE pseudoxanthoma elasticum "metabolic hypothesis" OR "metabolic disease" liver ABCC6 pyrophosphate parabiosis transplant evidence
pseudoxanthoma elasticum metabolic disease liver transplant parabiosis Abcc6
https://www.jci.org/articles/view/193499
"pseudoxanthoma elasticum" metabolic ABCC6 pyrophosphate inhibitor
https://pubmed.ncbi.nlm.nih.gov/18685618/
PMID: 28486967
The Metabolic Hypothesis of PXE
1. The Paradox That Generated the Hypothesis
| Hypothesis | Core claim |
|---|---|
| "PXE cell" hypothesis | Target-tissue cells (fibroblasts, smooth muscle cells) produce a defective matrix that mineralizes intrinsically |
| Metabolic hypothesis | A circulating anti-mineralization factor, whose production depends on hepatic ABCC6, is deficient systemically — so normal target-tissue cells mineralize because the blood bathing them lacks a protective factor |
2. The Experimental Proof: Transplantation and Parabiosis Studies
Skin Grafting (Jiang et al., J Invest Dermatol 2009 — PMID 18685618)
- Muzzle skin from wild-type (WT) mice was grafted onto the back of Abcc6−/− (knockout, KO) mice
- The WT skin underwent mineralization in the KO host
- KO muzzle skin grafted onto WT mice did not mineralize in the WT systemic environment
Conclusion: Mineralization is driven by the systemic (circulating) environment, not intrinsic properties of the skin cells. A protective factor present in WT blood is absent in KO blood.
Hepatocyte Transplantation
- Engraftment of WT hepatocytes into Abcc6−/− mice produced up to 90% reduction in muzzle skin mineralization vs. untreated controls
- Liver-specific deletion of Abcc6 was sufficient to cause post-injury cardiac calcification, while heart-specific deletion was not
Conclusion: The liver is the critical source of the circulating protective factor. Restoring hepatic ABCC6 function corrects the systemic deficiency.
Parabiotic Pairing (Jiang et al., Am J Pathol 2010)
- WT mice surgically joined to Abcc6−/− mice (sharing a common circulation)
- Mineralization in KO mice was halted
Conclusion: A diffusible blood-borne factor from the WT mouse continuously suppresses mineralization in the KO partner.
3. Identification of the Circulating Factor: Inorganic Pyrophosphate (PPi)
- ABCC6 on the basolateral membrane of hepatocytes mediates the efflux of ATP into the sinusoidal bloodstream
- The ectonucleotidase ENPP1 rapidly hydrolyzes extracellular ATP into AMP + inorganic pyrophosphate (PPi)
- PPi circulates as a potent inhibitor of hydroxyapatite crystal nucleation — it blocks calcium phosphate deposition onto organic matrices including elastic fibers
- In PXE, without hepatic ABCC6, ATP efflux is reduced → plasma PPi falls (~50% of normal) → mineralization goes unchecked
- Plasma PPi levels are measurably reduced in PXE patients
- ENPP1 mutations (GACI — Generalized Arterial Calcification of Infancy) cause a phenotypically overlapping disorder by disrupting the same pathway one step downstream
- Oral PPi supplementation reduces calcification in Abcc6−/− mice (Dedinszki et al., EMBO Mol Med 2017)
- Warfarin (which further impairs a secondary anti-mineralization pathway via vitamin K recycling and Matrix Gla Protein) dramatically accelerates mineralization in KO mice
4. The Complete Purinergic Pathway Model (2024)
Liver hepatocyte
↓ [ABCC6]
ATP efflux into portal circulation
↓ [ENPP1]
AMP + PPi ←— primary anti-mineralization factor
↓ [CD73 / NT5E]
Adenosine ←— secondary anti-mineralization molecule
↓ [TNAP] ←— consumes PPi (pro-calcification)
Pi (inorganic phosphate)
| Gene mutated | Disease | Step affected |
|---|---|---|
| ABCC6 | PXE | ATP efflux from liver |
| ENPP1 | GACI | ATP → PPi conversion |
| NT5E | CALJA | AMP → adenosine |
5. Challenges and Evolution of the Hypothesis (2025 Update)
- Tissue-specific Abcc6 deletion in the liver (but not in the heart) was sufficient to produce post-injury cardiac calcification
- Metabolomics and gene expression analysis showed deficiencies in nucleotide metabolism, cellular energetics, and mitochondrial respiration in affected tissues
- Evidence suggests ABCC6 may partly reside in mitochondria-associated ER membranes (MAMs), affecting mitochondrial function and lipid/calcium homeostasis
- Hepatic ABCC6 regulates mitochondrial function in distant organs through an altered systemic metabolic milieu — not only via PPi
The 2025 JCI data also explicitly notes that "pyrophosphate levels in individuals with PXE do not correlate well with the phenotype," suggesting PPi deficiency alone is not the complete explanation — broader metabolic disturbance (nucleotide pools, energy metabolism, mitochondrial function) contributes.
6. Clinical Corroboration: Acquired PXE After Liver Transplantation
- The disease can be transmitted through the liver (not through the skin or vessels)
- The liver is the driver of PXE pathology in humans, not just in mice
Summary: The Metabolic Hypothesis in One Paragraph
- Jiang Q et al. J Invest Dermatol 2009 (PMID 18685618) — original skin graft proof
- Germain DP. Orphanet J Rare Dis 2017 (PMID 28486967) — comprehensive review
- Kauffenstein G et al. Biology 2024 (PMID 38392293) — purinergic pathway model
- Pfau K et al. Prog Retin Eye Res 2024 (PMID 38815804) — genetics/pathophysiology review
- Dermatology 2-Volume Set 5e (Bologniaet al.) — "PXE represents a metabolic disorder with secondary connective tissue manifestations"
- JCI 2025 — liver-heart crosstalk via mitochondrial metabolic milieu
PXE and skin
pseudoxanthoma elasticum skin manifestations cutaneous lesions
pseudoxanthoma elasticum skin manifestations cutaneous features histology 2024 2025
https://pmc.ncbi.nlm.nih.gov/articles/PMC4561240/
pseudoxanthoma elasticum skin papules neck flexural histology calcified elastic fibers
PXE and the Skin
1. Why the Skin Is Affected: The Tissue Basis
2. Clinical Cutaneous Features
2a. Primary Lesions
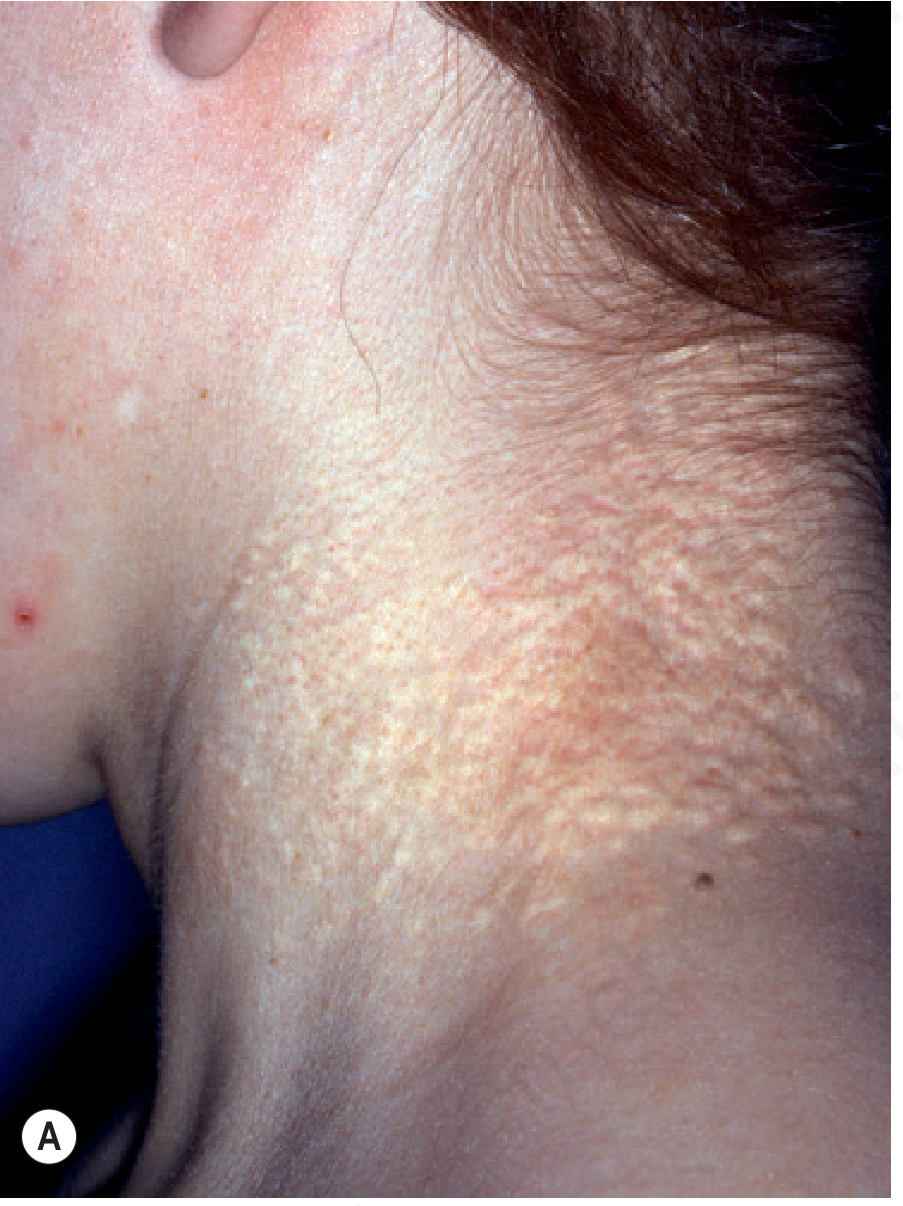
- "Plucked chicken skin" — the classic descriptor; the neck skin looks like the skin of a plucked bird
- "Cobblestone" appearance — as individual papules coalesce
- "Peau d'orange" texture — dimpled, orange-peel-like surface at a later stage
2b. Sites of Predilection
| Site | Comment |
|---|---|
| Lateral neck | Most consistent, earliest, most visible |
| Axillae | Frequently involved |
| Antecubital and popliteal fossae | Classical flexural sites |
| Inguinal folds | Often affected |
| Periumbilical region | Distinctive involvement |
| Periauricular skin | Commonly overlooked |
| Mental creases (chin) | Horizontal chin creases before age 30 are highly suggestive of PXE |
| Nasolabial folds | Characteristically exaggerated |
2c. Progressive Changes
- Lax and pendulous — due to loss of elastic fiber structural integrity
- Thickened and leathery — especially in advanced disease
- Redundant skin folds — most prominent on the lateral neck and axillae
2d. Mucosal Involvement
- Soft palate
- Inner lip
- Tonsils
- Stomach, rectum, and vagina (detected on endoscopy/biopsy)
2e. Nuchal Comedones and Milia en Plaque
3. Histopathology of the Skin
3a. Primary Findings (Light Microscopy)
| Feature | Detail |
|---|---|
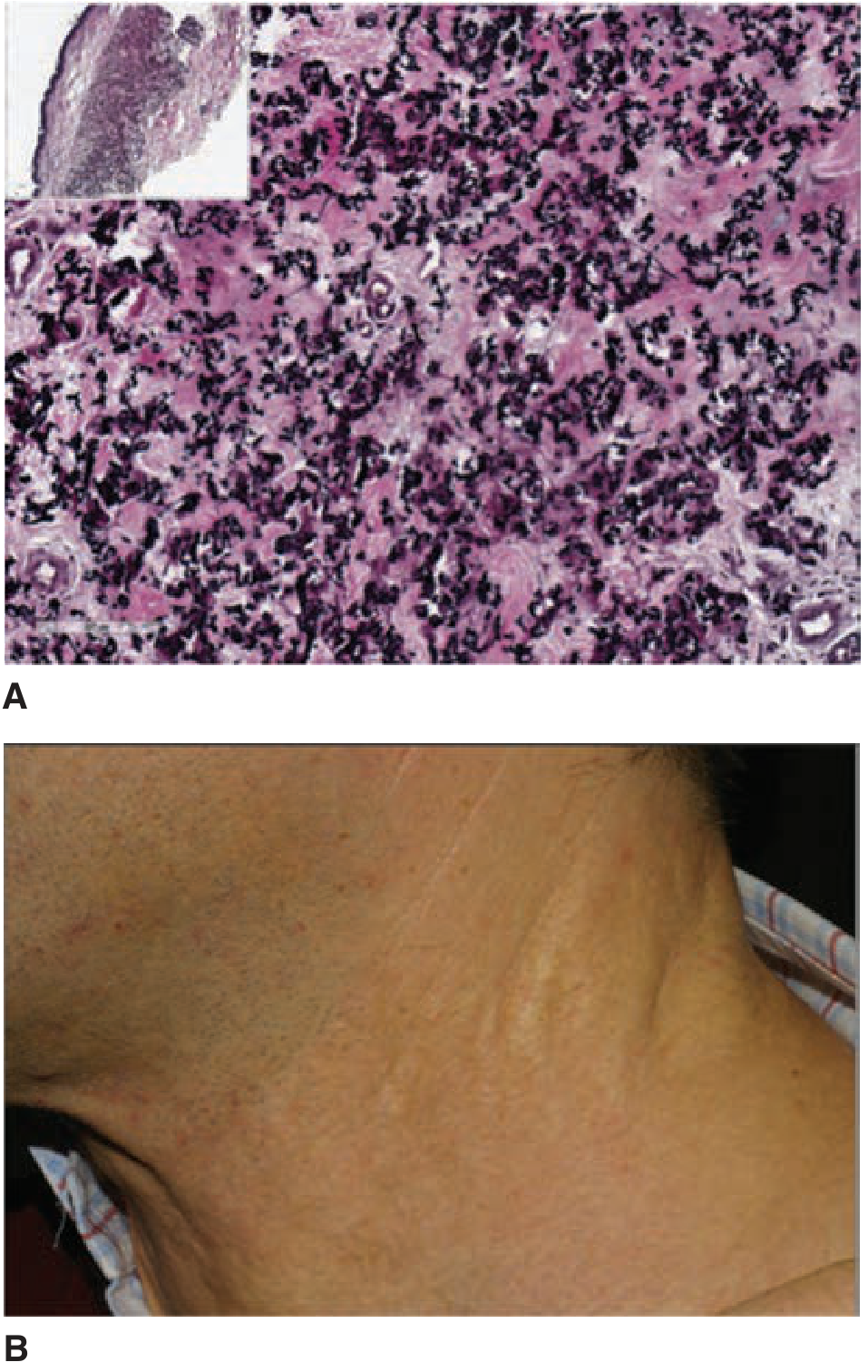
| Elastorrhexis | Fragmented, short, twisted, curled elastic fibers — the primary histological hallmark |
| Calcified elastic fibers | Mid-to-deep dermis; calcium deposits on the elastic fibers ("mineralized elastin") |
| H&E staining | Elastic fibers appear gray-blue, twisted and broken — described as "raveled wool" |
| Distribution | Mid and lower dermis; the upper (papillary) dermis is typically spared |
| Collagen fibers | Collagen flowers (abnormal fibrils) and increased proteoglycans may be seen adjacent to mineralized fibers |
3b. Special Stains
| Stain | Result / Use |
|---|---|
| Von Kossa | Black deposits — confirms calcium (most sensitive for early disease) |
| Alizarin red | Red/orange deposits — calcium |
| Verhoeff–van Gieson (VVG) | Highlights elastic fiber fragmentation |
| Calcium stains | Helpful for early/subclinical disease in normal-appearing skin |
3c. Ultrastructure (Electron Microscopy)
- Elastic fibers show pleomorphic mineralized deposits within the amorphous elastin core and along microfibrils
- Calcium crystals (hydroxyapatite) are oriented along the long axis of fibers
- Fibroblasts are hypertrophied with prominent rough ER (suggesting reactive synthetic activity)
- Macrophages are abundant within calcified deposits (phagocytosing mineral)
- Ultrastructural elastic fiber degeneration can be found in clinically non-lesional skin — important for early/blind biopsies
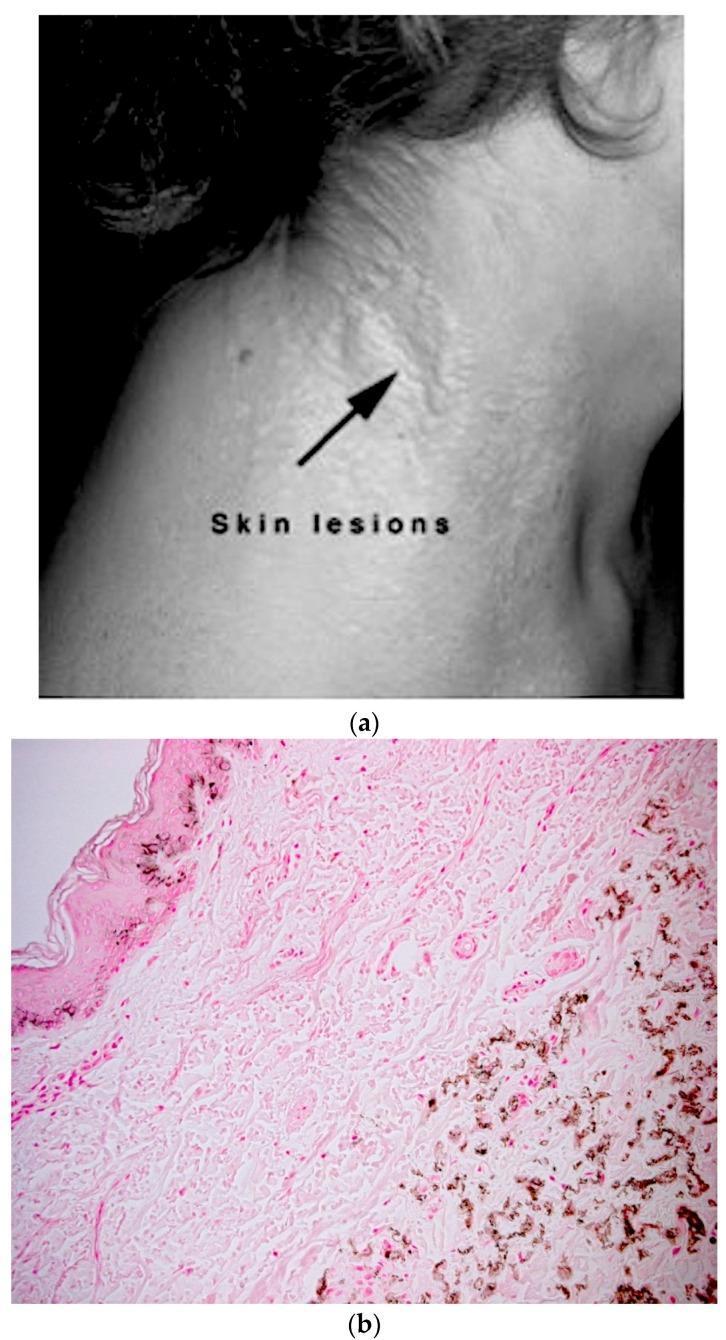
4. Diagnostic Significance of the Skin
Skin as Diagnostic Gateway
- Both major skin criteria plus one major eye criterion, or
- Major genetic criterion (biallelic pathogenic ABCC6 variants)
- Characteristic pseudoxanthomatous papules and plaques on the neck and/or flexural sites
- Histopathologic evidence of calcified elastic fibers in the mid and lower dermis of affected skin
Diagnostic Biopsy Pearls
- Blind biopsies of old scars or axillary skin in patients with a family history or angioid streaks may reveal early PXE changes even without clinical skin lesions
- Non-lesional skin biopsy showing histopathologic PXE changes = minor criterion
- Mental creases before age 30 are highly suggestive and should prompt biopsy
5. PXE-Like Skin Conditions: Differential Diagnosis
| Condition | Distinguishing features |
|---|---|
| PXE-like papillary dermal elastolysis | Cobblestone yellow papules on neck; NO retinal/vascular disease; NO calcification on histology |
| Elastosis perforans serpiginosa | Red serpiginous/annular papules; transepidermal elimination of elastic material |
| Cutis laxa | Skin laxity without papules; elastic fibers reduced not calcified |
| Connective tissue nevus with elastorrhexis | Small white papules, upper chest/neck; no systemic findings |
| Perforating calcific elastosis (Kyrle's) | Periumbilical in multiparous, obese women; associated with chronic renal disease |
| D-penicillamine–induced PXE-like changes | Drug history (Wilson's, homocystinuria); NO calcification; lesions may resolve after stopping drug |
| β-thalassemia / sickle cell disease | Up to 20% develop PXE-like skin; no ABCC6 mutation; due to downregulation of ABCC6 expression in liver |
| GGCX mutations | PXE-like skin + vitamin K-dependent coagulation factor deficiency |
| Amyloid elastosis | Amyloid deposits encasing dermal elastic fibers; in setting of primary systemic amyloidosis |
| Acanthosis nigricans | Brown velvety plaques; different morphology; metabolic/insulin association |
Key rule: No calcification on histology → most likely NOT true PXE. No retinal/vascular findings with only skin changes → consider PXE-like papillary dermal elastolysis.
6. Skin as Biomarker and Clinical Signpost
- Their presence signals ongoing elastic fiber calcification throughout the body
- Severity of skin involvement does not reliably predict ocular or cardiovascular severity (significant intrafamilial variability even among siblings)
- Appearance of mental creases or nuchal skin changes in a young person (<30 years) should trigger ophthalmologic assessment and cardiovascular screening
- Gastric involvement (mucosa) explains the ~10% risk of gastric hemorrhage
7. Treatment of Skin Manifestations
- Cosmetic/surgical: Plastic surgery for redundant skin folds (particularly neck and axillary) in severe cases
- Dietary: Limiting dietary calcium and phosphorus to the minimum daily requirement — some evidence of benefit
- Emerging systemic therapies targeting the underlying mineralization (PPi analogues/etidronate, TNAP inhibitors, recombinant ENPP1, magnesium supplementation, inositol, sodium thiosulfate) may indirectly slow cutaneous progression
- A randomized controlled trial of etidronate (a PPi analogue) showed it may halt progression of arterial calcification; data on skin is less established
- Dermatology 2-Volume Set 5e (Bolognini et al.) — Ch. 97, §§ Clinical Features, Pathogenesis, Diagnostic Criteria, Differential Diagnosis
- Andrews' Diseases of the Skin — Ch. 25, PXE
- Boraldi F et al. Int J Mol Sci 2015 — "PXE and skin: Clinical manifestations, histopathology, pathomechanism, perspectives of treatment" (PMID 26361562)
- Cleveland Clinic Journal of Medicine 2026 — cobblestone appearance, bramble-bush histology
give some pictures of the cosmetic plastic surgery, target ectopic mineralization, etidronate, TNAP inhibitors, recombinant ENPP1, Magnesium, sodium thiosulfate, inositol