I now have comprehensive material to deliver a thorough answer. Let me compile everything:
Myocardial Infarction (MI) - Comprehensive Review
Definition
Myocardial infarction (MI) - commonly called a "heart attack" - is necrosis of cardiac muscle resulting from prolonged ischemia. The 2018 universal definition requires the presence of acute myocardial injury (detected by abnormal cardiac biomarkers) in the setting of evidence of acute myocardial ischemia.
Approximately 800,000 individuals in the United States experience an MI each year (nearly one MI every 40 seconds), causing almost 400,000 deaths annually.
- Robbins & Kumar Basic Pathology, p. 467
Epidemiology & Risk Factors
-
~10% of MIs occur before age 40; 45% occur before age 65
-
Risk rises progressively with age and atherosclerotic risk factors
-
Males have higher relative risk through middle age
-
Females are generally protected during reproductive years, but postmenopausal decline in estrogen (with rising cholesterol, BP, and inflammatory markers) erases this protection - IHD is the most common cause of death in older women
-
Postmenopausal hormone replacement therapy has not been shown to be protective, and may be detrimental (prothrombotic effect)
-
Robbins, Cotran & Kumar Pathologic Basis of Disease, p. 511
Pathogenesis
The Typical Sequence (responsible for ~90% of MIs)
- An atheromatous plaque is eroded or suddenly disrupted by endothelial injury, intraplaque hemorrhage, or mechanical forces - exposing subendothelial collagen and necrotic plaque contents
- Platelets adhere, aggregate, and are activated, releasing thromboxane A₂, ADP, and serotonin - causing further platelet aggregation and vasospasm
- Coagulation is activated by exposure of tissue factor, adding to the growing thrombus
- Within minutes, the enlarging thrombus completely occludes the coronary artery lumen
Angiography performed within 4 hours of MI onset demonstrates coronary thrombosis in almost 90% of cases. By 12-24 hours (without intervention), only 60% show thrombosis - meaning some occlusions clear spontaneously.
- Robbins & Kumar Basic Pathology, p. 468-469
Causes When Typical Atherothrombosis is Absent (~10% of cases)
- Coronary vasospasm (with or without atherosclerosis; cocaine, ephedrine)
- Embolism - from left atrial mural thrombus (AF), infective endocarditis vegetations, prosthetic material, or paradoxical embolism via patent foramen ovale
- Small vessel disease - vasculitis, amyloid deposition, sickle cell disease
- Prolonged demand-supply mismatch (tachycardia/hypertension in setting of fixed stenosis) - causing subendocardial necrosis without thrombus
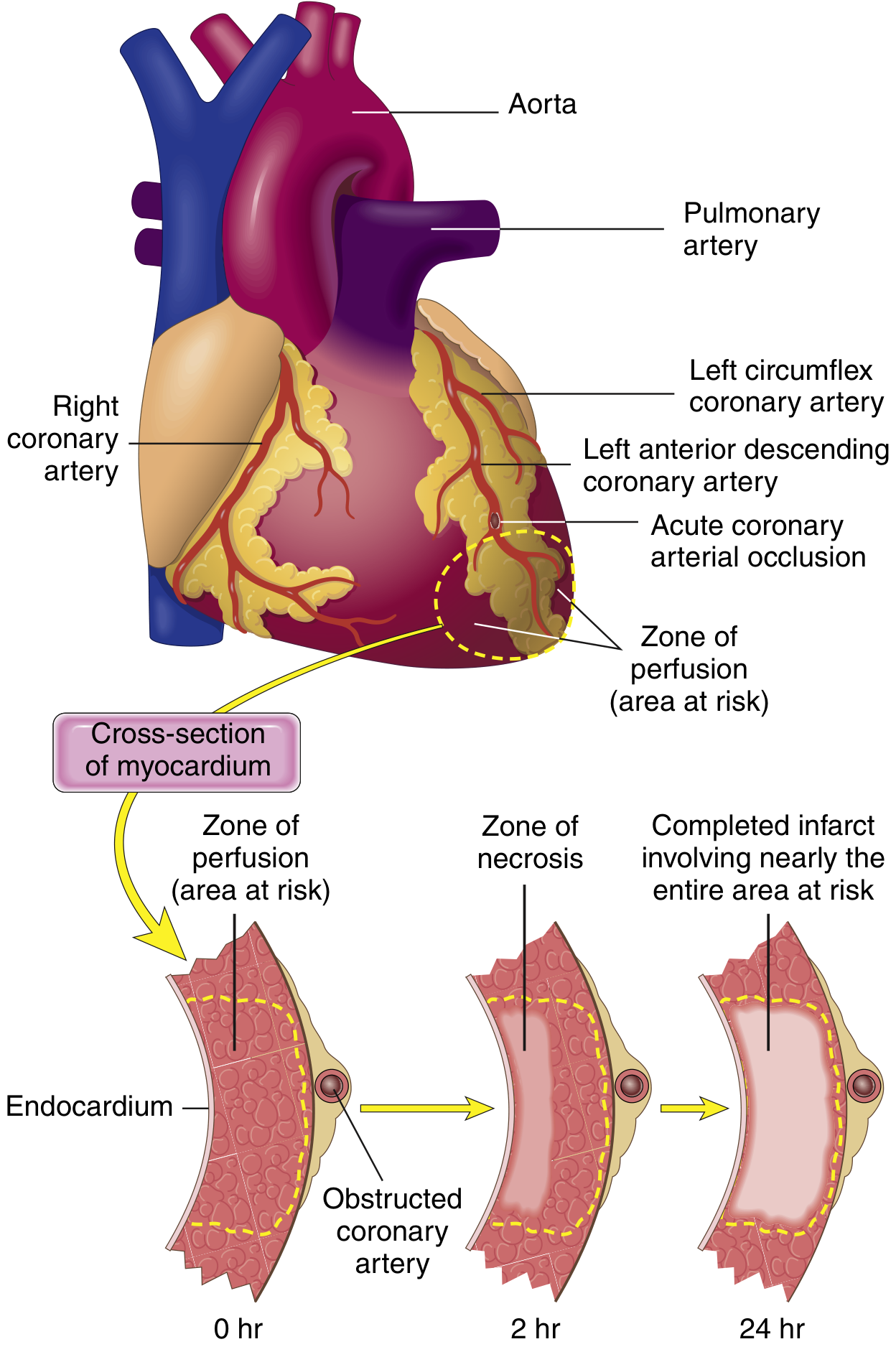
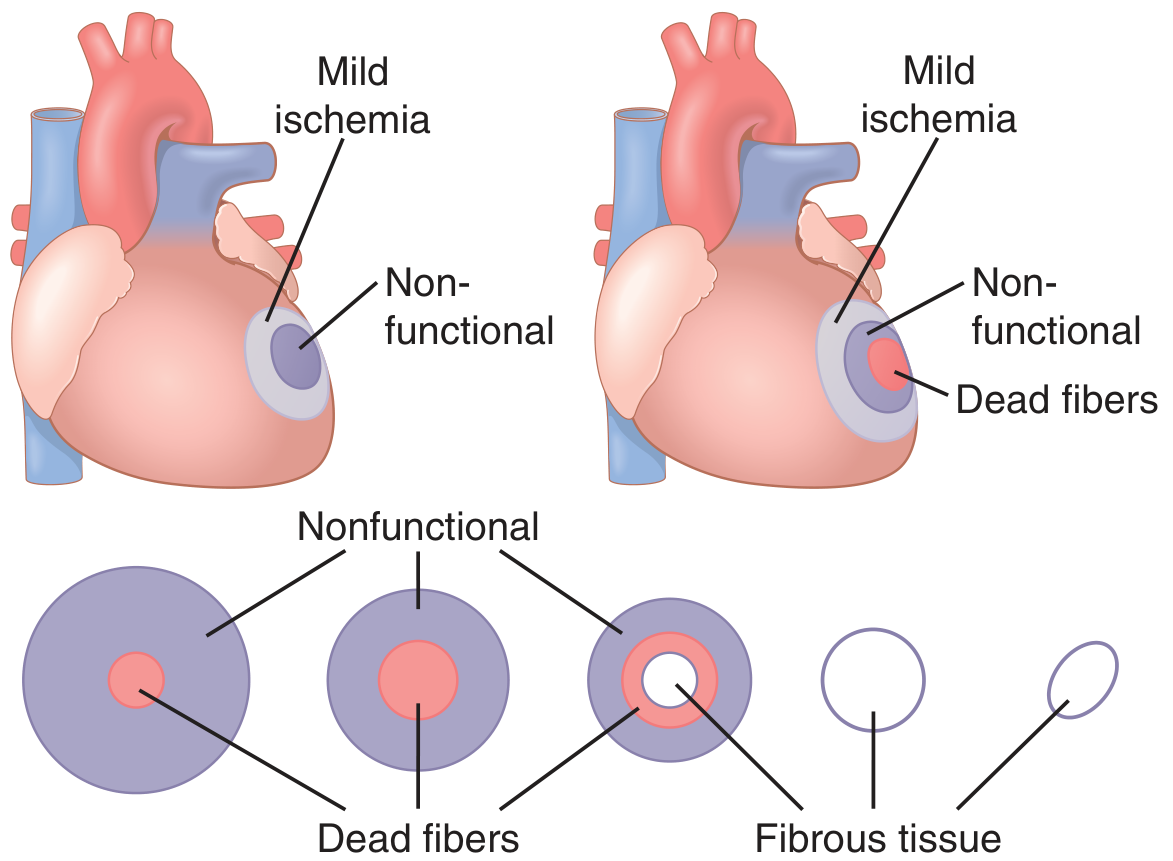
Progression of Necrosis: The Wavefront Phenomenon
FIG. 9.8 from Robbins Basic Pathology - necrosis begins in the subendocardial region and expands as a wavefront toward the epicardium, achieving full extent in 3-6 hours.
-
The subendocardium is most vulnerable because it is furthest from epicardial vessels and has the highest intramural pressures impairing blood flow
-
Cardiac muscle requires ~1.3 mL O₂/100 g/min just to remain alive; normal resting delivery is ~8 mL O₂/100 g/min
-
Even 15-30% of normal collateral flow can prevent cell death in border zones; however, the central zone of a large infarct has almost zero flow and does die
-
Guyton & Hall Textbook of Medical Physiology, p. 271
Infarct Patterns by Coronary Artery
| Vessel | Frequency | Territory Infarcted |
|---|
| LAD (proximal) | 40-50% | Anterior LV wall, anterior 2/3 of septum, apex |
| RCA (proximal) | 30-40% | Most of RV, posterior septum (in right-dominant heart), inferior LV |
| LCX (proximal) | 15-20% | Lateral LV wall |
- Robbins & Kumar Basic Pathology, p. 537
Morphological Changes Over Time
The following table summarizes the classic sequence (from Robbins Basic Pathology, Table 9.2):
| Time Frame | Gross Features | Light Microscopy | EM Findings |
|---|
| 0-30 min | None | None | Relaxation of myofibrils; glycogen loss; mitochondrial swelling |
| 30 min - 4 hrs | None | Usually none; variable waviness of fibers at border | Sarcolemmal disruption; mitochondrial amorphous densities |
| 4-12 hrs | Occasionally dark mottling | Onset coagulation necrosis; edema; hemorrhage | - |
| 12-24 hrs | Dark mottling | Ongoing coagulation necrosis; pyknotic nuclei; hypereosinophilic myocytes; marginal contraction band necrosis; early neutrophilic infiltrate | - |
| 1-3 days | Mottling with yellow-tan center | Loss of nuclei and striations; interstitial infiltration by neutrophils | - |
| 3-7 days | Hyperemic border; central yellow-tan softening | Phagocytosis of dead cells by macrophages; risk of rupture highest | - |
| 7-10 days | Maximally yellow-tan and soft | Well-developed granulation tissue at margins | - |
| 2-8 weeks | Gray-white scar, progressing inward | Fibrosis | - |
| >2 months | Complete scarring | Dense collagenous scar | - |
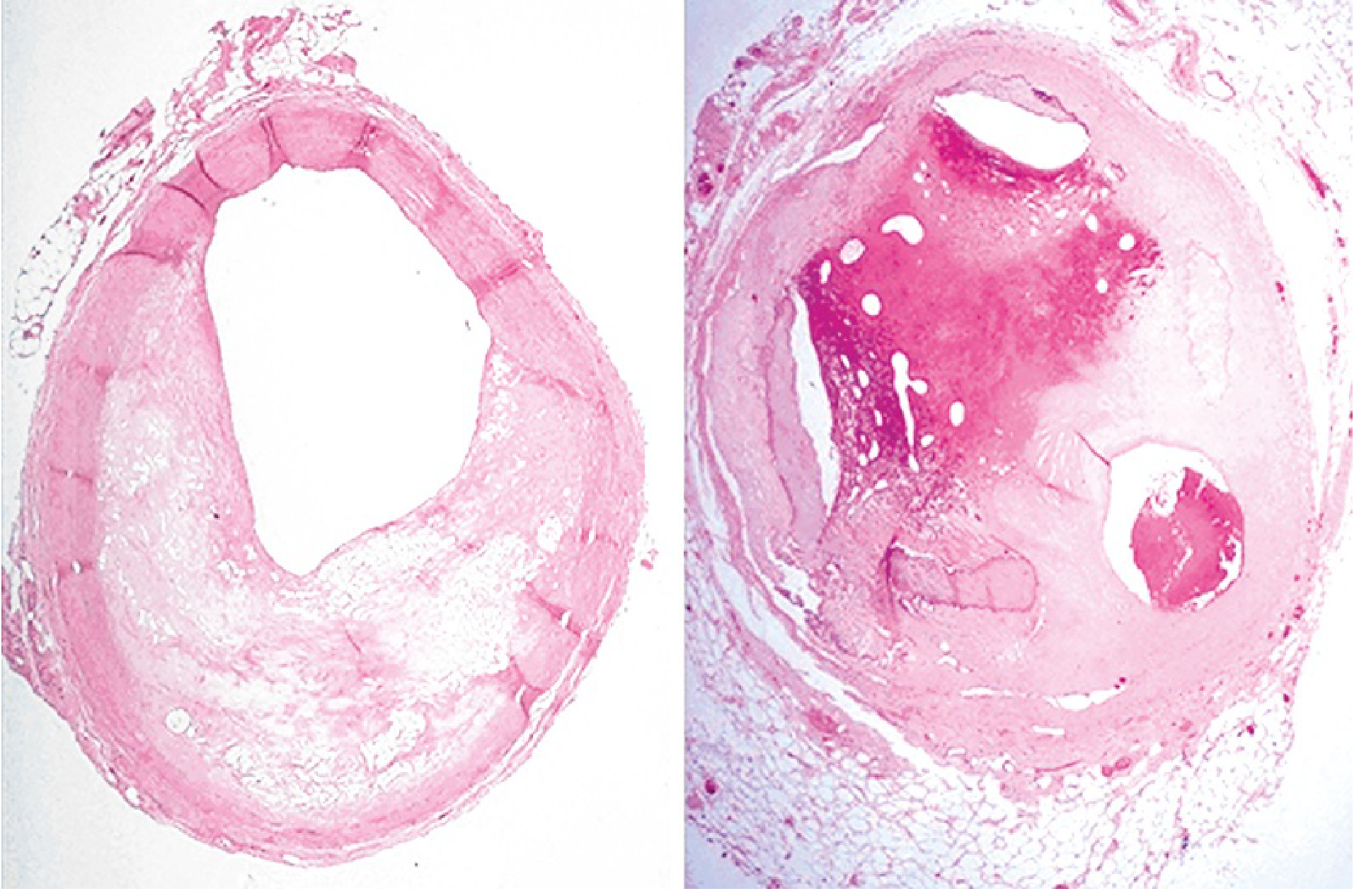
Key point: Very recent infarcts (<12 hours) are often not grossly apparent. Triphenyl tetrazolium chloride (TTC) staining (a substrate for LDH) identifies infarcts >3 hours old - the infarcted area appears pale/unstained because LDH leaks out of necrotic cells.
Coronary atherosclerosis (Robbins Basic Pathology, eFig. 9.1) - left: 60-70% stenosis; right: near-complete occlusion from previous thrombosis with partial organization.
Biochemistry of Ischemia
Within seconds of vascular obstruction:
- Aerobic metabolism ceases
- ATP drops, lactic acid accumulates
- Contractility is lost within minutes (reversible at this stage)
After 20-40 minutes of ischemia:
- Irreversible damage begins
- Sarcolemmal integrity is lost - intracellular macromolecules (troponin, CK-MB) leak into bloodstream
In 80-90% of cases, cardiac death in ischemia is due to ventricular fibrillation (from electrical instability of ischemic myocardium), not mechanical failure.
- Robbins & Kumar Basic Pathology, p. 491-493
Cardiac Biomarkers
The hallmark diagnostic test. Key markers and their timing:
| Biomarker | Rises | Peaks | Normalizes | Notes |
|---|
| Troponin I / T | 3-6 hrs | 24-48 hrs | 5-14 days | Most sensitive/specific; preferred marker |
| CK-MB | 3-12 hrs | 24 hrs | 2-3 days | Useful for re-infarction detection |
| Myoglobin | 1-4 hrs | 6-12 hrs | 24 hrs | Earliest rise but not cardiac-specific |
| LDH | 24 hrs | 3-6 days | 8-14 days | Historically used; less specific |
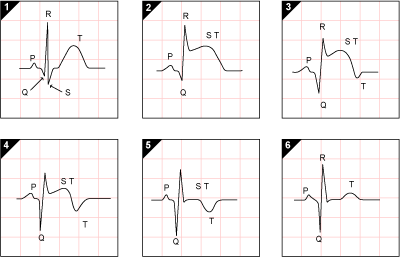
ECG Changes
Three major mechanisms cause ECG changes in acute MI (Ganong's Review of Medical Physiology):
| Defect in Infarcted Cells | Current Flow | ECG Change |
|---|
| Rapid repolarization (accelerated K⁺ channel opening) | Out of infarct | ST elevation |
| Decreased resting membrane potential (loss of intracellular K⁺) | Into infarct | TQ depression (manifested as ST elevation) |
| Delayed depolarization | Out of infarct | ST elevation |
Evolution of ECG changes:
- Acute (hours): Hyperacute T waves → ST elevation (STEMI) or ST depression (NSTEMI/subendocardial)
- Days-weeks: ST segment returns toward baseline; T wave inversions appear
- Weeks-months: Pathological Q waves develop in leads over the infarct area (dead tissue is electrically silent)
Leads on the opposite side of the heart show reciprocal ST depression.
Clinical Presentation
Classic: Severe crushing/pressure-like central chest pain, radiating to the left arm, jaw, or back; associated with diaphoresis, nausea, dyspnea, and sense of impending doom. Pain lasts >30 minutes and is not relieved by nitrates.
Atypical presentations (more common in women, diabetics, elderly):
- Epigastric pain or heartburn-like discomfort
- Jaw or arm pain alone
- Dyspnea without chest pain
- Fatigue, syncope
- "Silent MI" (especially in diabetics with autonomic neuropathy)
Causes of Death After MI
From Guyton & Hall Textbook of Medical Physiology:
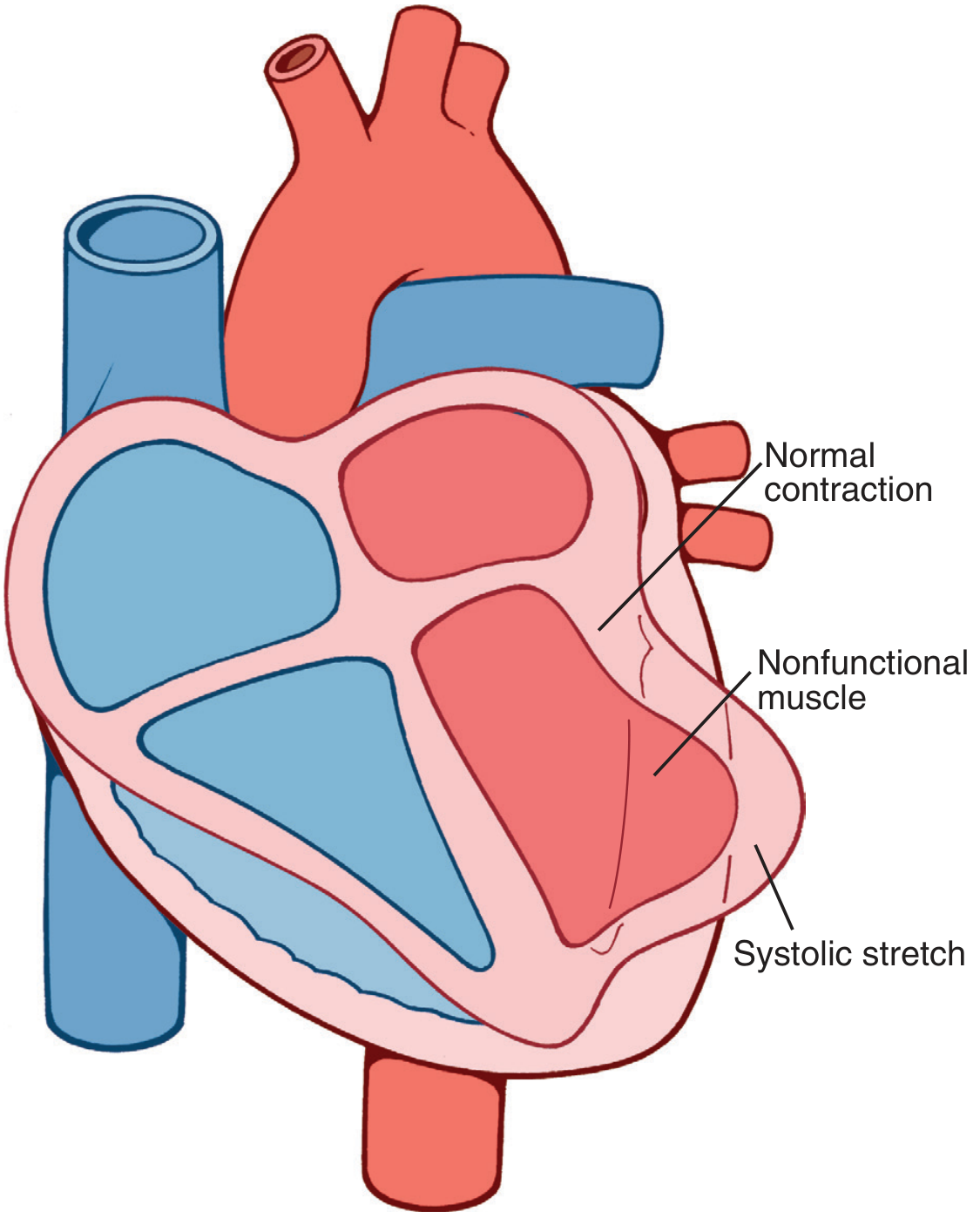
- Decreased cardiac output - infarcted muscle fails to contract; systolic stretch (dyskinesis) worsens this, as normal myocardium pushes ischemic segments outward (paradoxical motion), wasting contractile energy
- Pulmonary edema - from left ventricular failure and elevated filling pressures
- Ventricular fibrillation - most common cause of death (80-90% of ischemic cardiac deaths)
- Cardiac rupture - free wall rupture (days 3-7, peak risk), papillary muscle rupture (acute MR), ventricular septal rupture
Complications
| Complication | Timing | Mechanism |
|---|
| Arrhythmias (VF, VT, heart block) | Hours to days | Electrical instability of ischemic/necrotic tissue |
| LV failure / cardiogenic shock | Acute | Loss of >40% of LV mass |
| Pericarditis (fibrinous) | Days 2-4 | Necrosis extending to epicardium |
| Free wall rupture | Days 3-7 | Neutrophil-mediated lysis at weakest point |
| VSR (ventricular septal rupture) | Days 3-7 | Same mechanism; inferior MI (RCA) predisposed |
| Papillary muscle rupture | Days 2-7 | Posteromedial papillary muscle (single blood supply from RCA) most vulnerable → acute severe MR |
| LV aneurysm (true) | Weeks-months | Scarring + ventricular remodeling |
| Mural thrombus | Days-weeks | Stasis over akinetic LV wall → risk of systemic embolism |
| Dressler syndrome | 2-10 weeks | Autoimmune pericarditis |
Management
Immediate (STEMI)
The goal is rapid reperfusion. Time = myocardium.
- Aspirin 160-325 mg chewed immediately (buccal absorption; inhibits COX-1/thromboxane A₂)
- O₂ only if SpO₂ <90%; routine O₂ not recommended with normal saturations
- Nitroglycerin sublingual 0.4 mg q5 min (up to 3 doses) - reduces preload and dilates coronary vessels
- Avoid if: SBP <90 mmHg, RV infarction (inferior ECG changes + JVP elevated + clear lungs), or phosphodiesterase-5 inhibitor use within 24 hours
- Morphine 2-4 mg IV q5 min for pain relief (vagotonic - watch for bradycardia/hypotension)
- Beta-blockers (e.g., metoprolol 5 mg IV q2-5 min, 3 doses) - reduce O₂ demand, decrease VF risk, reduce reinfarction; avoid if HR <60, SBP <100, or >1st degree heart block
- Primary PCI - preferred reperfusion if achievable within 120 min of first medical contact
- Fibrinolysis - if PCI not available within 120 min
- Anticoagulation - heparin, enoxaparin, or bivalirudin
- Dual antiplatelet therapy - aspirin + P2Y₁₂ inhibitor (clopidogrel, ticagrelor, or prasugrel)
- Harrison's Principles of Internal Medicine 22E, p. 1201-1213
Long-term Secondary Prevention
- Beta-blocker (reduces mortality post-MI, though recent meta-analysis data (PMID: 39298680) questions benefit in patients with preserved EF without heart failure)
- ACE inhibitor/ARB (especially with reduced EF, anterior MI, or heart failure)
- Statin (high-intensity)
- Dual antiplatelet therapy for 12 months
- Aldosterone antagonist (eplerenone/spironolactone) in post-MI with EF ≤40% and heart failure or diabetes
Recent Evidence (2024-2026)
- Beta-blockers post-MI with preserved EF: A 2025 meta-analysis in Eur J Prev Cardiol (PMID: 39298680) questions the long-held practice of universal beta-blocker use after MI in patients without reduced EF or heart failure - suggesting limited benefit in this subgroup.
- Sudden cardiac death post-MI: A 2024 pooled cohort meta-analysis in Eur Heart J (PMID: 39378245) updated risk stratification data for SCD after MI.
Key Mnemonics
MONA (classic initial treatment): Morphine - Oxygen - Nitrates - Aspirin
(Note: Current guidelines de-emphasize routine oxygen and morphine should be used selectively)
Reperfusion injuries to remember: Contraction band necrosis, arrhythmias (reperfusion VF), microvascular injury ("no-reflow"), myocardial stunning
Sources: Robbins & Kumar Basic Pathology; Robbins, Cotran & Kumar Pathologic Basis of Disease; Harrison's Principles of Internal Medicine 22E; Guyton & Hall Textbook of Medical Physiology; Ganong's Review of Medical Physiology; Goldman-Cecil Medicine; Fuster & Hurst's The Heart