I have my university exam of 1 st year of subject biochemistry These are the important long answer give me answer of this "Glycine Metabolism" and remeber it's a university examination
Reading File
Finding Sources
Finding Sources
Reading File
Finding Sources
Reading File
Finding Sources
Reading File
Reading File
Reading File
Loading Image
Loading Image
Finding Sources
I now have comprehensive content from multiple authoritative biochemistry textbooks. Let me compile the full university-level long answer.
Glycine Metabolism
(University Examination - 1st Year Biochemistry)
Introduction
Glycine (H2N-CH2-COOH) is the simplest amino acid - it has only a hydrogen atom as its side chain, making it the only amino acid that is NOT optically active (no chiral center). It is a non-essential, glucogenic amino acid. Despite its structural simplicity, glycine participates in a wide variety of metabolic pathways.
1. Structure of Glycine
- Molecular formula: C2H5NO2
- Simplest amino acid; no chiral carbon
- Non-essential (body can synthesize it)
- Glucogenic amino acid (its catabolism produces intermediates that enter gluconeogenesis)
2. Synthesis (Biosynthesis) of Glycine
A. From Serine (Major Route)
The primary route of glycine synthesis is the reversible conversion of serine to glycine, catalyzed by serine hydroxymethyltransferase (also called glycine hydroxymethyltransferase).
Serine + Tetrahydrofolate (FH4) → Glycine + N5,N10-methylene-FH4
- Coenzymes required: Tetrahydrofolate (FH4) and Pyridoxal Phosphate (PLP)
- In this reaction, the hydroxymethyl group of serine is transferred to FH4, forming N5,N10-methylene-FH4 (a one-carbon donor)
- The reaction is reversible - it is the major route for BOTH synthesis of glycine AND its conversion back to serine
B. From Threonine (Minor Route)
Threonine can undergo an aldolase-like cleavage (because threonine has a hydroxyl group two carbons from the carbonyl) to yield glycine and acetaldehyde. This is a minor pathway in humans.
Threonine → Glycine + Acetaldehyde (catalyzed by threonine aldolase, requires PLP)
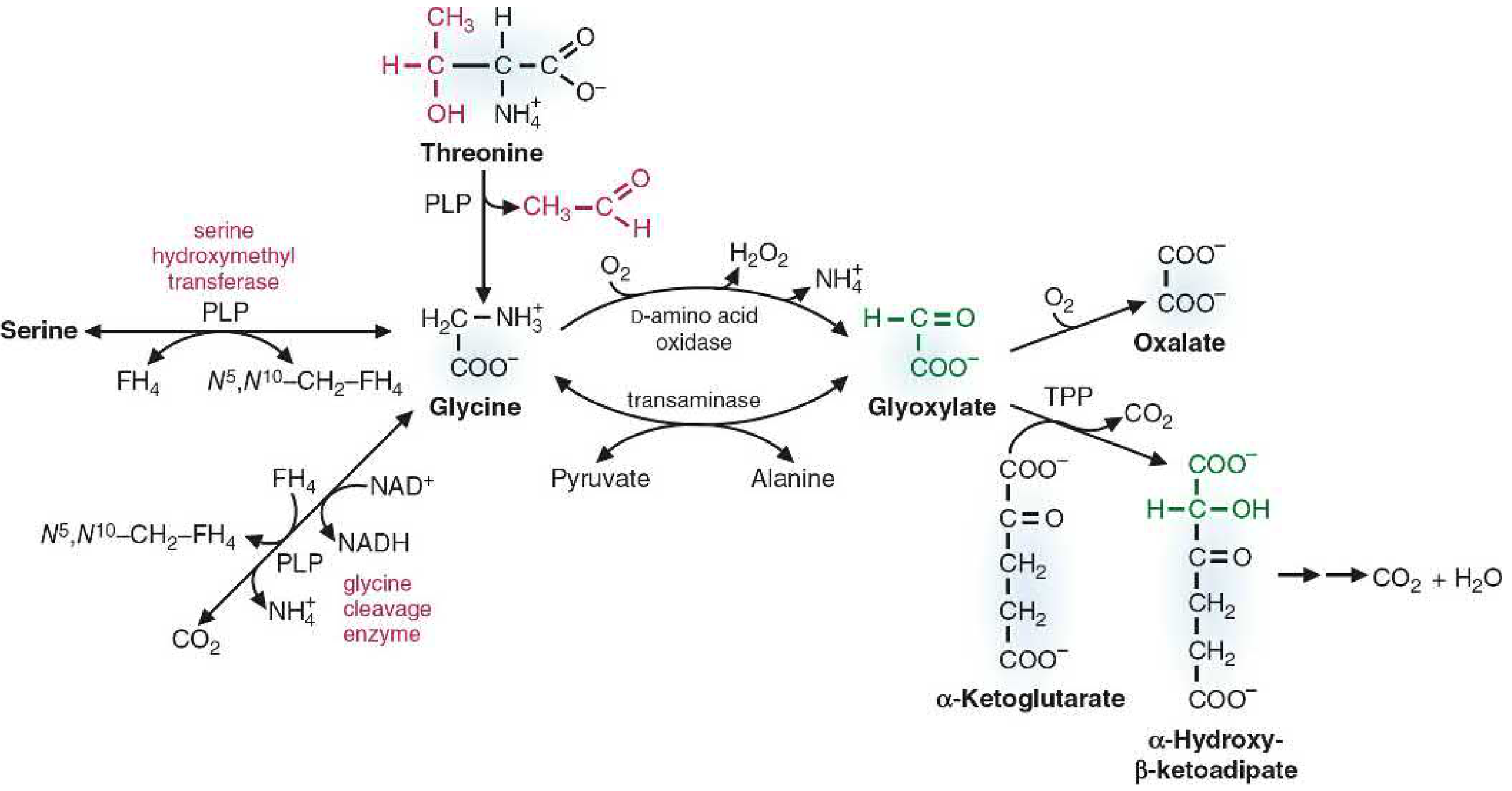
3. Metabolism (Degradation/Catabolism) of Glycine
Glycine can be catabolized by multiple pathways:
A. Glycine Cleavage System (Major Degradative Route)
This is the primary catabolic pathway for glycine. It occurs in liver mitochondria and is catalyzed by the glycine cleavage complex (also called glycine decarboxylase complex).
Overall reaction:
Glycine + FH4 + NAD+ → CO2 + NH3 + N5,N10-methylene-FH4 + NADH + H+
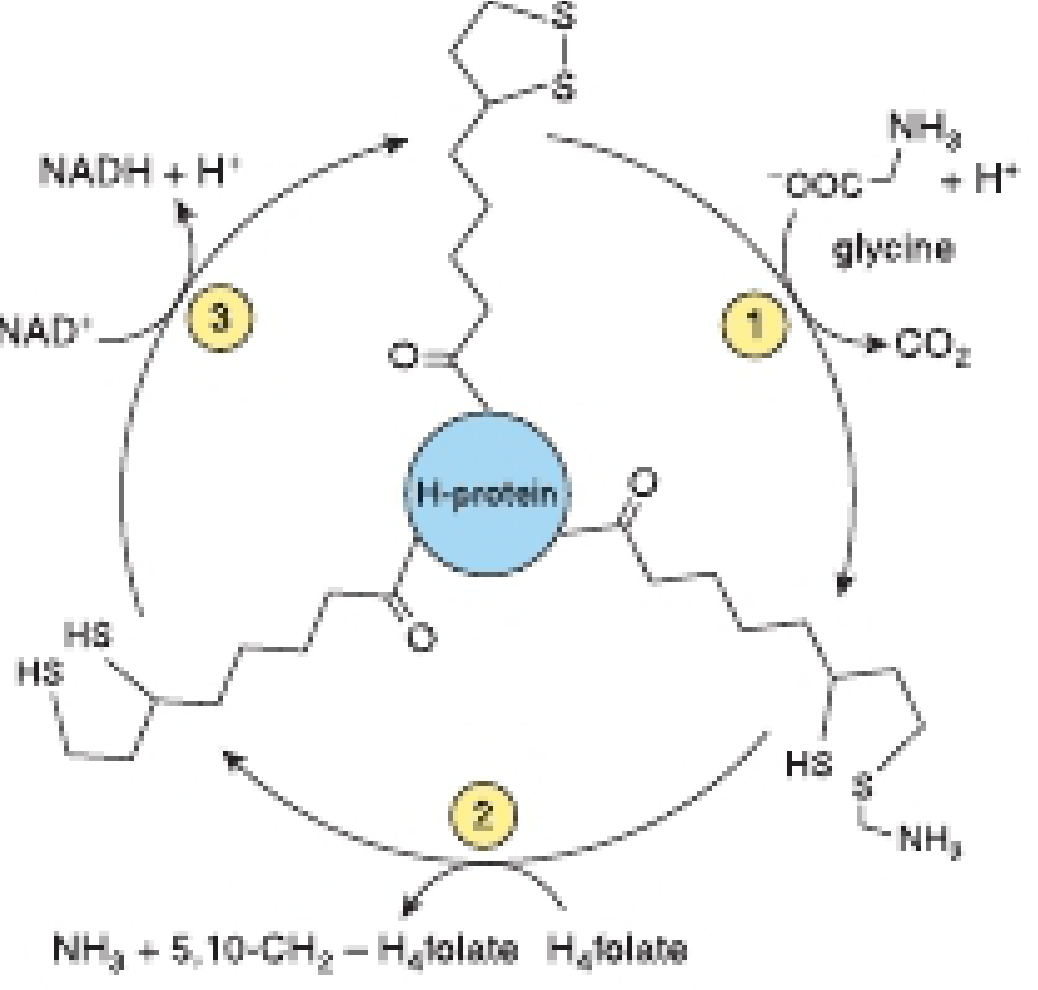
Components of the Glycine Cleavage Complex:
The complex consists of three enzymes and a scaffold protein:
| Component | Function |
|---|---|
| P-protein (Glycine dehydrogenase, decarboxylating) | Step 1: Decarboxylates glycine, releasing CO2; amino group transferred to H-protein |
| H-protein | Carrier protein with a covalently attached dihydrolipoyl moiety; acts as a swinging arm to carry intermediates between enzyme active sites |
| T-protein (Aminomethyltransferase) | Step 2: Transfers the aminomethyl group from H-protein to FH4, releasing NH3 and forming N5,N10-methylene-FH4 |
| L-protein (Dihydrolipoamide dehydrogenase) | Step 3: Reoxidizes the dihydrolipoyl group on H-protein using NAD+, producing NADH |
Significance: The N5,N10-methylene-FH4 produced can be used in one-carbon transfer reactions (e.g., thymidylate synthesis, purine biosynthesis).
B. Conversion Back to Serine
Since the serine hydroxymethyltransferase reaction is reversible:
Glycine + N5,N10-methylene-FH4 → Serine + FH4
This is an important anabolic route when serine is needed.
C. Glyoxylate Pathway
Glycine can be deaminated to form glyoxylate by the enzyme D-amino acid oxidase (a flavoprotein):
Glycine + O2 → Glyoxylate + H2O2 + NH4+
Glyoxylate can then be further metabolized:
-
Oxidation to oxalate:Glyoxylate + O2 → Oxalate (insoluble)Oxalate forms insoluble calcium salts → deposits in kidney tubules → kidney stones (renal calculi). About 40% of oxalate in the liver is derived from glycine catabolism.
-
Transamination back to glycine:Glyoxylate + Alanine → Glycine + Pyruvate (requires a peroxisomal transaminase)
-
Condensation with alpha-ketoglutarate (via TPP): forms alpha-hydroxy-beta-ketoadipate → CO2 + H2O
4. Metabolic Roles of Glycine (Anabolic Functions)
Glycine is a building block for many important biomolecules:
| Product | Role of Glycine |
|---|---|
| Porphyrins / Heme | Glycine + Succinyl-CoA → delta-Aminolevulinic acid (ALA), the first committed step in heme synthesis |
| Purines | Glycine contributes carbons C-4, C-5, and N-7 of the purine ring |
| Creatine | Glycine + Arginine → Guanidinoacetate → Creatine (with methionine as methyl donor) |
| Glutathione | Glycine is one of the three amino acids (Glu-Cys-Gly) that form glutathione |
| Bile acid conjugation | Glycine conjugates with bile acids (cholic acid, chenodeoxycholic acid) to form glycocholate and glycochenodeoxycholate; increases their water solubility |
| Hippuric acid | Glycine conjugates with benzoic acid in liver to form hippuric acid (a detoxification product) |
| Collagen | Glycine constitutes every 3rd residue in collagen (Gly-X-Y repeat) |
| One-carbon metabolism | Glycine-serine interconversion donates one-carbon units to FH4 for methylation reactions |
| Sarcosine (N-methylglycine) | Under high methionine/SAM conditions, glycine can be methylated to sarcosine; sarcosine is then catabolized back to glycine |
5. Role in One-Carbon Metabolism
The serine-glycine interconversion is tightly linked to folate-mediated one-carbon metabolism:
- Serine → Glycine donates a one-carbon unit (methylene group) to tetrahydrofolate (FH4), forming N5,N10-methylene-FH4
- This activated one-carbon unit is used in:
- Thymidylate synthesis (dTMP from dUMP - essential for DNA replication)
- Purine biosynthesis
- Methionine regeneration (via methylcobalamin and methionine synthase)
6. Clinical Disorders of Glycine Metabolism
A. Non-Ketotic Hyperglycinemia (NKH) / Glycine Encephalopathy
- Cause: Defect in the glycine cleavage system (most commonly the P-protein or T-protein)
- Inheritance: Autosomal recessive
- Result: Glycine accumulates in ALL body tissues, especially the CNS
- Features: Neonatal onset of seizures, hypotonia, lethargy, apnea, intellectual disability; often fatal in infancy
- Glycine acts as an inhibitory neurotransmitter in the spinal cord and an excitatory co-agonist at NMDA receptors in the brain - excess glycine disrupts both functions
B. Primary Hyperoxaluria Type 1 (PH1)
- Cause: Deficiency of the peroxisomal enzyme alanine:glyoxylate aminotransferase (AGT) in the liver
- Result: Glyoxylate (from glycine catabolism) cannot be transaminated back to glycine; instead it is oxidized to oxalate
- Features: Recurrent kidney stones (calcium oxalate), nephrocalcinosis, progressive renal failure, early mortality from hypertension/renal failure
C. Glycinuria
- Cause: Defect in renal tubular reabsorption of glycine
- Result: Excess glycine excreted in urine (generally benign)
7. Summary Diagram of Glycine Metabolism
Glucose (3-Phosphoglycerate)
↓
Serine ←————————————→ Glycine ←——— Threonine (minor)
↑ Serine ↑
(FH4-PLP) hydroxy- FH4-PLP
methyltransferase
Glycine metabolic fates:
1. → Serine (reversible, via FH4) — one-carbon donation
2. → CO2 + NH3 (Glycine Cleavage System in mitochondria, via FH4, NAD+)
3. → Glyoxylate (D-amino acid oxidase)
→ Oxalate (→ kidney stones if excess)
→ Back to Glycine (transaminase)
→ CO2 + H2O (via α-ketoglutarate)
Biosynthetic roles:
Glycine → Heme (+ Succinyl-CoA)
Glycine → Purines (C4, C5, N7)
Glycine → Creatine
Glycine → Glutathione
Glycine → Bile acid conjugates
Glycine → Hippuric acid (detox)
Glycine → Collagen (every 3rd residue)
8. Key Enzymes and Coenzymes - Summary Table
| Reaction | Enzyme | Coenzyme |
|---|---|---|
| Serine → Glycine | Serine hydroxymethyltransferase | PLP, FH4 |
| Glycine → CO2 + NH3 | Glycine cleavage complex (P, H, T, L proteins) | PLP, FH4, NAD+, Lipoic acid |
| Glycine → Glyoxylate | D-amino acid oxidase | FAD (flavoprotein) |
| Glyoxylate → Glycine | Alanine:glyoxylate aminotransferase (AGT) | PLP |
| Glyoxylate → α-hydroxy-β-ketoadipate | Glyoxylate transaminase | TPP |
| Glycine + Succinyl-CoA → ALA | ALA synthase | PLP |
Key Points to Remember for Exam
- Glycine is the simplest and only non-chiral amino acid
- Synthesized primarily from serine (via serine hydroxymethyltransferase; PLP + FH4)
- Major catabolism: glycine cleavage system in liver mitochondria - 3 enzymes + H-protein; produces CO2, NH3, N5,N10-methylene-FH4, NADH
- Glyoxylate pathway → oxalate → kidney stones (primary hyperoxaluria type 1 = AGT deficiency)
- Glycine is a precursor of heme, purines, creatine, glutathione, bile salts, collagen
- Key disorder: Non-ketotic hyperglycinemia = glycine cleavage system defect → glycine accumulation → seizures, encephalopathy
- Glycine is a glucogenic amino acid (carbons enter gluconeogenesis via pyruvate or other intermediates)
Sources: Basic Medical Biochemistry - A Clinical Approach, 6e (Lieberman & Peet); Harper's Illustrated Biochemistry, 32e; Lippincott Illustrated Reviews: Biochemistry, 8e
This is a shared conversation. Sign in to Orris to start your own chat.