Uric acid metabolism from Harrison for 10 marks question f
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
I now have everything needed for a comprehensive 10-mark answer. Here is the full answer based directly on Harrison's Principles of Internal Medicine, 22nd Edition (2025):
Uric Acid Metabolism
(Harrison's Principles of Internal Medicine, 22nd Ed — Chapter 384)
1. End Product of Purine Metabolism
Uric acid is the final catabolic end product of purine metabolism in humans. At physiologic pH (7.4), it exists predominantly as urate (the ionized form), given its weak acidic property (pKa = 5.8). This is unique to humans: most other mammals possess uricase (urate oxidase), which converts urate into the highly water-soluble compound allantoin. Humans (and some higher primates) have an evolutionary loss of functional uricase, making them uniquely susceptible to hyperuricemia and gout.
2. Pathways of Urate Production
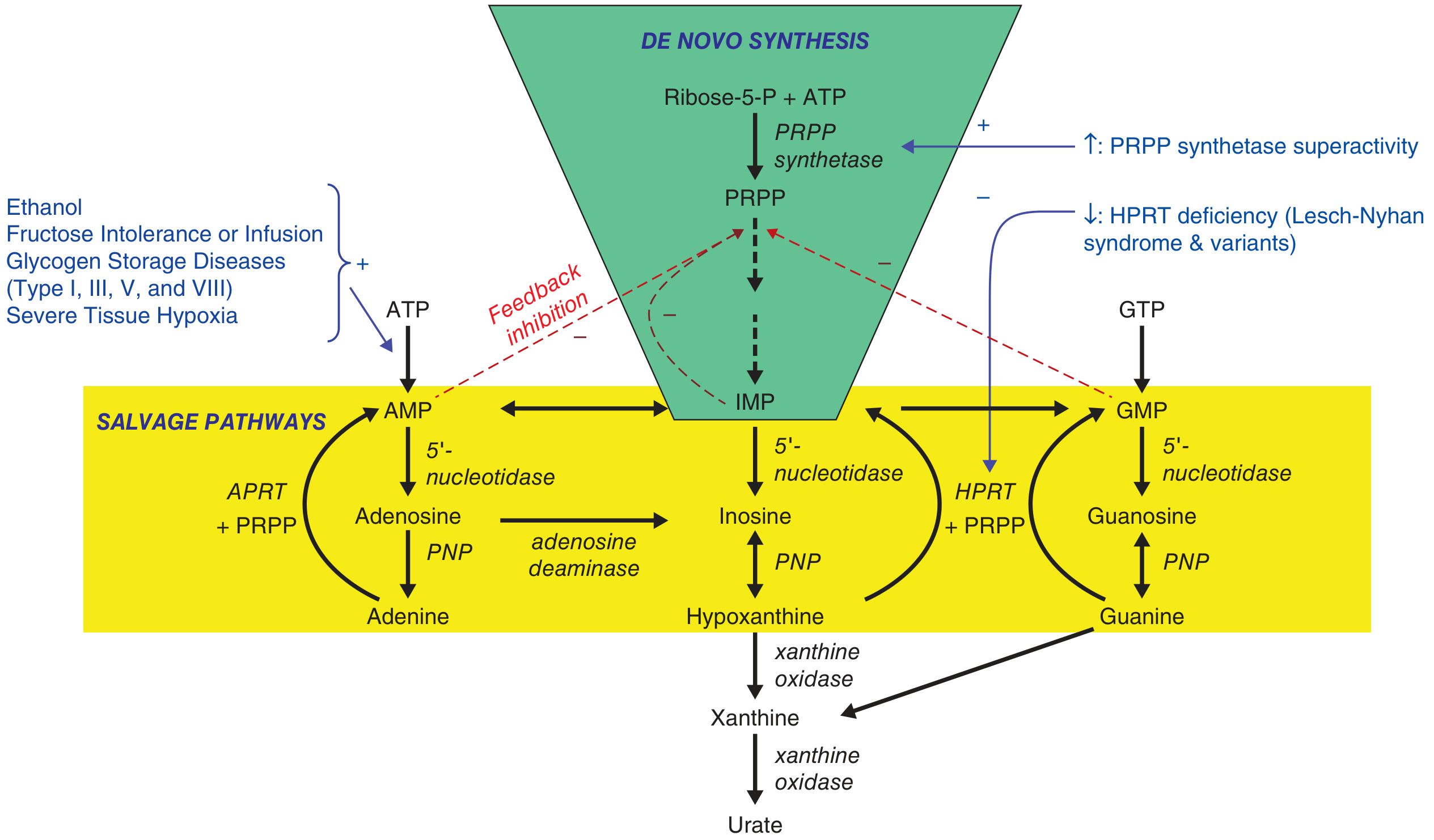
There are two main routes through which purines are synthesised, ultimately yielding urate:
A. De Novo Synthesis
- Starts with Ribose-5-phosphate + ATP, converted by PRPP synthetase (PRS) to 5-phosphoribosyl-1-pyrophosphate (PRPP)
- PRPP is the key branch-point substrate funnelled into the purine ring synthesis pathway
- End products of the pathway are AMP, IMP, and GMP (purine nucleotides)
- These are degraded sequentially to nucleosides → free bases (hypoxanthine, guanine) → xanthine → urate via xanthine oxidase
B. Salvage Pathway
- Free purine bases released from tissue nucleic acid turnover are "salvaged" (recycled) back to nucleotides
- HPRT (hypoxanthine-guanine phosphoribosyltransferase) salvages:
- Hypoxanthine → Inosine monophosphate (IMP)
- Guanine → Guanosine monophosphate (GMP)
- APRT (adenine phosphoribosyltransferase) salvages adenine → AMP
- The salvage pathway reduces net urate production by recycling purines — its failure (HPRT deficiency) leads to overproduction
C. Final Steps — Xanthine Oxidase
- Hypoxanthine → Xanthine (via xanthine oxidase)
- Xanthine → Urate (via xanthine oxidase)
- Guanine is also converted to xanthine via guanine deaminase
- Allopurinol and febuxostat inhibit xanthine oxidase, blocking the final steps
3. Regulation and Feedback
- IMP, AMP, and GMP provide feedback inhibition of PRPP synthetase and the de novo pathway, controlling the rate of purine synthesis
- When HPRT is absent or deficient (as in Lesch-Nyhan syndrome), salvage is lost → PRPP is not consumed in the salvage pathway → PRPP accumulates → drives de novo synthesis → massive urate overproduction
4. Causes of Urate Overproduction
| Mechanism | Example |
|---|---|
| PRPP synthetase superactivity | X-linked inborn error; leads to ↑PRPP → ↑purine nucleotides → ↑urate |
| HPRT deficiency (partial) | Kelley-Seegmiller syndrome → gout + urate nephropathy |
| HPRT deficiency (complete) | Lesch-Nyhan syndrome → self-mutilation + hyperuricemia |
| ↑ATP degradation to AMP → urate | Alcohol (↑NAD reduction), fructose ingestion, glycogen storage diseases (Types I, III, V, VIII), severe tissue hypoxia |
| Increased cell turnover | Myeloproliferative disorders, psoriasis, hemolytic anemia, cytotoxic therapy |
5. Urate Excretion
The systemic urate concentration reflects a balance between dietary intake, endogenous synthesis, and excretion:
- ~2/3 of daily uric acid is eliminated via the kidney (renal excretion)
- ~1/3 via the gastrointestinal tract (intestinal uricolysis by gut bacteria)
Renal Handling (Proximal Tubule)
A four-component model operates in the proximal tubule:
- Filtration at glomerulus
- Near-complete reabsorption (~98–99%) early proximal tubule
- Secretion mid-proximal tubule
- Post-secretory reabsorption
Key transporters:
- URAT1 and OAT10 (apical membrane) — mediate urate reabsorption via sodium-coupled anion exchange; inhibited by uricosuric drugs (probenecid, losartan, SGLT2 inhibitors)
- GLUT9 (basolateral membrane) — exits urate into interstitium
- ABCG2 — high-capacity urate efflux transporter in renal proximal tubule AND intestinal epithelium; genetic variants are the most common cause of impaired extra-renal urate excretion
Anti-uricosuric agents (e.g., pyrazinoate, nicotinate, low-dose aspirin) promote urate reabsorption via these transporters, raising serum urate.
6. Hyperuricemia: Mechanisms
Hyperuricemia (serum urate >6.85 mg/dL) arises from:
- Impaired renal excretion — accounts for ~90% of cases (diuretics, CKD, lead nephropathy, cyclosporine)
- Overproduction — ~10% (inborn errors, myeloproliferative disorders, high purine diet)
- Combined mechanisms — most cases of clinical gout
7. Pathogenesis of Gout from Hyperuricemia
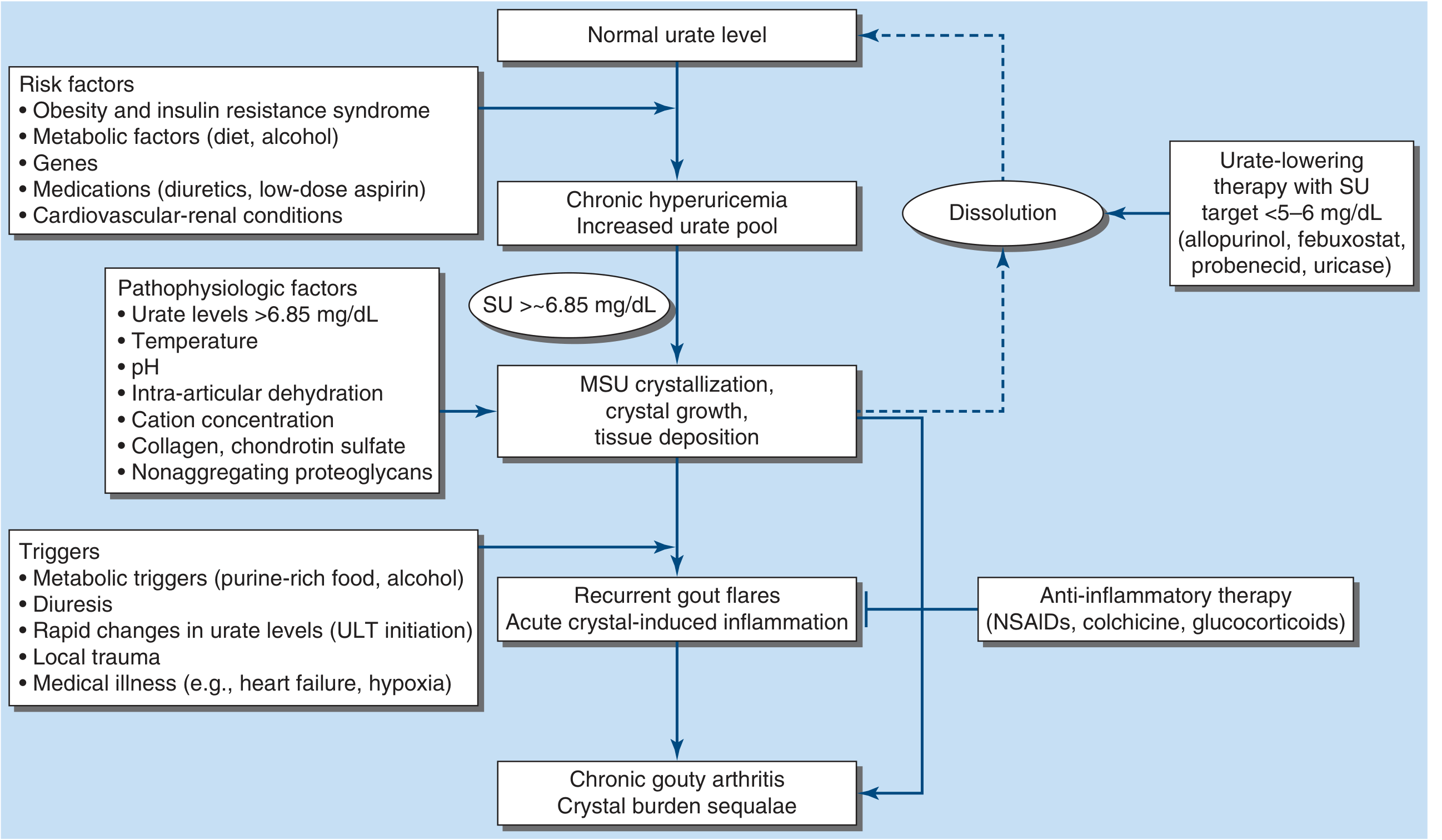
Chronic hyperuricemia → supersaturation → monosodium urate (MSU) crystal deposition in joints and connective tissue → phagocytosis by neutrophils → NLRP3 inflammasome activation → IL-1β, IL-6, TNF-α release → acute gouty arthritis. Persistent crystal deposition leads to tophi and chronic gouty arthritis.
8. Normal Values and Measurement
- Serum uric acid: 238–516 μmol/L (4.0–8.7 mg/dL) by uricase method
- Useful to diagnose gout and monitor urate-lowering therapy (target: <5–6 mg/dL for dissolution of crystals)
Summary Table
| Step | Enzyme / Transporter | Clinical Relevance |
|---|---|---|
| De novo PRPP synthesis | PRPP synthetase | Superactivity → ↑urate |
| Salvage of hypoxanthine/guanine | HPRT | Deficiency → Lesch-Nyhan; ↑urate |
| Hypoxanthine → Xanthine → Urate | Xanthine oxidase | Target of allopurinol, febuxostat |
| Renal reabsorption | URAT1, OAT10 | Target of probenecid, SGLT2i |
| Basolateral exit | GLUT9 | Key transporter |
| Intestinal excretion | ABCG2 | Variants → underexcretion gout |
— Harrison's Principles of Internal Medicine, 22nd Edition (2025), Chapter 384: Crystal-Induced Arthropathies
Aerobic glycolysis from Harrison and standard biochemistry harperfor 10 marks question and answer with relevant good flowcharts for final yr pg general medicine
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Loading Image
I now have all three textbook sources and the key diagrams. Let me compile the complete 10-mark answer:
Aerobic Glycolysis — 10 Mark Answer
(Sources: Harper's Illustrated Biochemistry 32nd Ed, Basic Medical Biochemistry 6th Ed, Harrison's Principles of Internal Medicine 22nd Ed)
1. Definition and Significance
Aerobic glycolysis is the cytosolic, oxygen-dependent pathway in which one molecule of glucose (6-carbon) is oxidized to two molecules of pyruvate (3-carbon each), generating ATP, NADH, and biosynthetic precursors, with oxygen required to reoxidize the NADH produced.
- Location: Cytosol (all enzymes are cytosolic)
- Glucose is the universal fuel for human cells — the brain uses it almost exclusively; after a high-carbohydrate meal, glucose is the major fuel for all tissues
- Aerobic glycolysis sets the stage for complete oxidation via the TCA cycle + oxidative phosphorylation, yielding ~30–32 mol ATP per mol glucose
2. Two Phases of Glycolysis
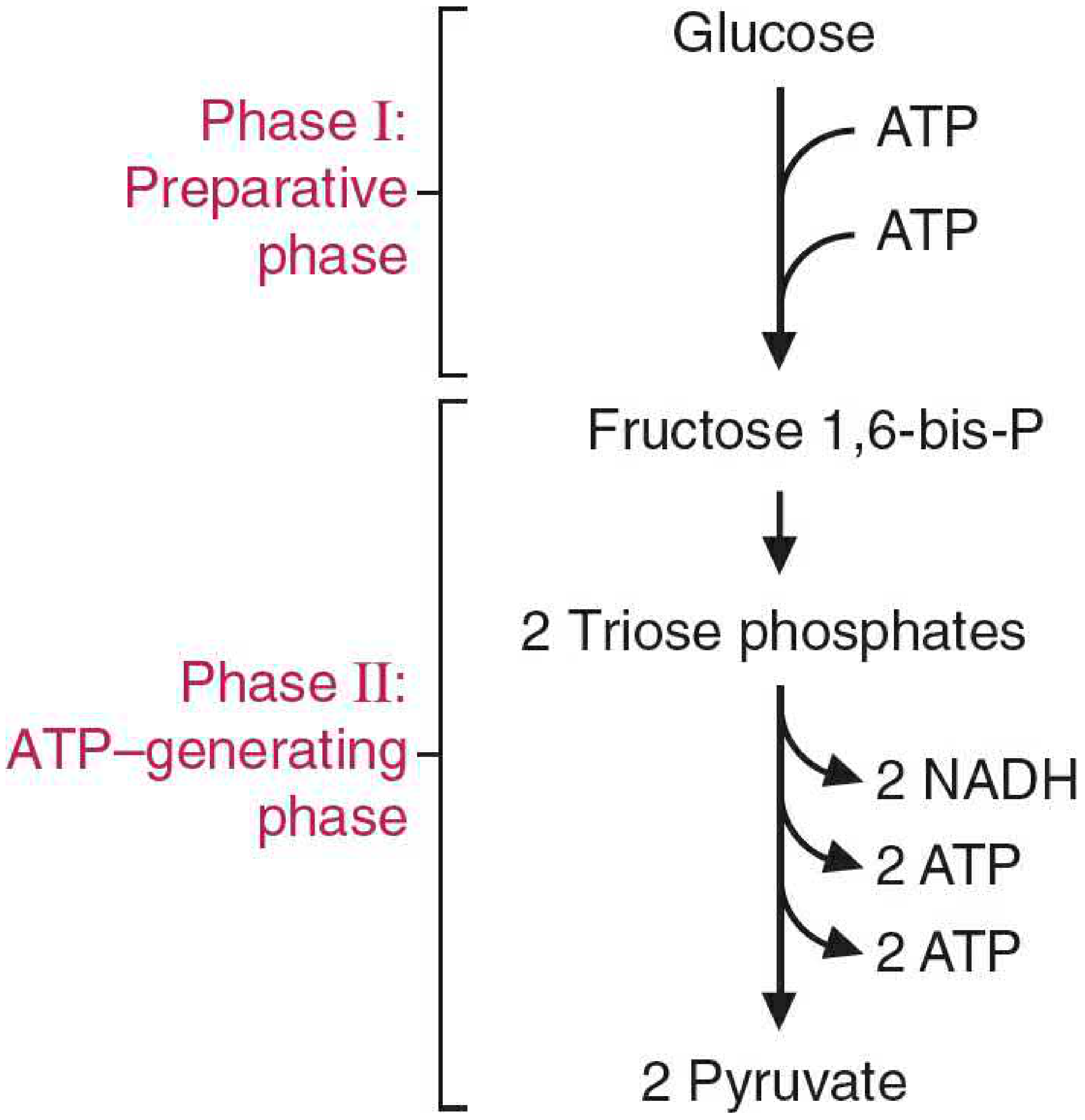
| Phase | Description | ATP change |
|---|---|---|
| Phase I — Preparatory ("priming") | Glucose → Fructose-1,6-bisphosphate | −2 ATP (invested) |
| Phase II — ATP-generating | 2× Triose phosphate → 2× Pyruvate | +4 ATP + 2 NADH |
| Net | +2 ATP + 2 NADH |
3. The 10 Reactions of Glycolysis (Harper's Fig. 17-2)
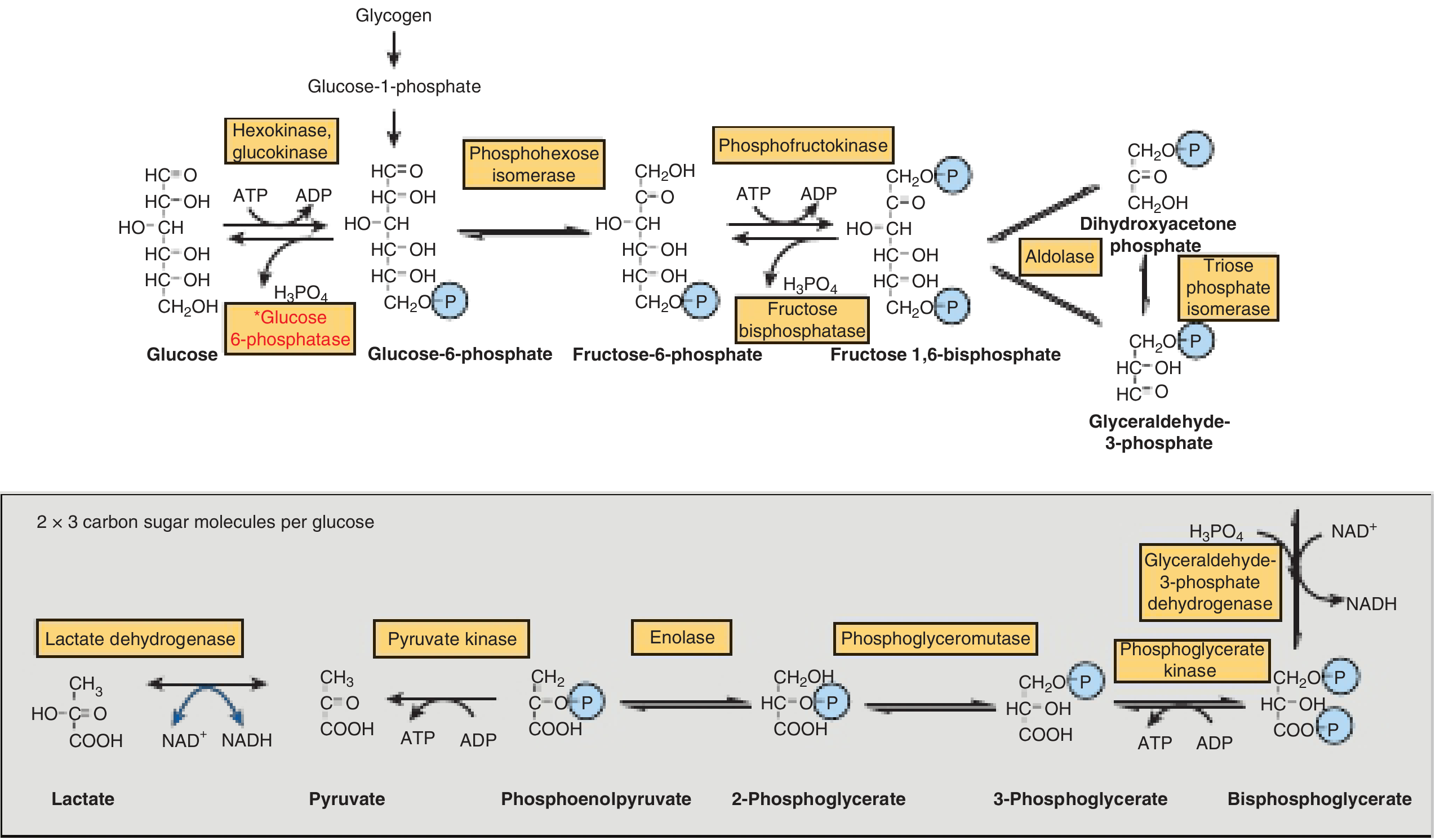
Phase I — Preparatory Phase (Reactions 1–5)
| Step | Reaction | Enzyme | Notes |
|---|---|---|---|
| 1 | Glucose → Glucose-6-phosphate (G6P) | Hexokinase (all tissues) / Glucokinase (liver, pancreatic β-cells) | ATP consumed; irreversible; HK inhibited by G6P; GK not inhibited by G6P, high Km |
| 2 | G6P → Fructose-6-phosphate (F6P) | Phosphoglucose isomerase | Reversible; isomerization |
| 3 | F6P → Fructose-1,6-bisphosphate (F1,6-BP) | Phosphofructokinase-1 (PFK-1) | ATP consumed; rate-limiting, committed step; irreversible |
| 4 | F1,6-BP → DHAP + Glyceraldehyde-3-phosphate (G3P) | Aldolase | Cleavage of 6C → two 3C molecules |
| 5 | DHAP ⇌ G3P | Triose phosphate isomerase | Equilibrium; DHAP converted to G3P for further metabolism |
From this point, all reactions occur twice (×2) per glucose
Phase II — ATP-Generating Phase (Reactions 6–10)
| Step | Reaction | Enzyme | Notes |
|---|---|---|---|
| 6 | G3P + NAD⁺ + Pi → 1,3-Bisphosphoglycerate (1,3-BPG) | G3P dehydrogenase | Generates 2 NADH; high-energy acyl-phosphate bond formed; inhibited by arsenate |
| 7 | 1,3-BPG + ADP → 3-Phosphoglycerate (3-PG) + ATP | Phosphoglycerate kinase | Substrate-level phosphorylation (2 ATP); "pays back" the investment |
| 8 | 3-PG → 2-Phosphoglycerate (2-PG) | Phosphoglycerate mutase | Phosphate shifted from C3→C2 |
| 9 | 2-PG → Phosphoenolpyruvate (PEP) | Enolase | Dehydration; generates high-energy enol-phosphate; inhibited by fluoride |
| 10 | PEP + ADP → Pyruvate + ATP | Pyruvate kinase | Substrate-level phosphorylation (2 ATP); irreversible; inhibited by ATP, alanine; activated by F1,6-BP (feedforward) |
4. Summary: Overall Equation
Aerobic glycolysis:
Glucose + 2 NAD⁺ + 2 Pi + 2 ADP → 2 Pyruvate + 2 NADH + 2 H⁺ + 2 ATP + 2 H₂O
ΔG°' ≈ −22 kcal/mol (pathway is overall irreversible)
5. Fate of Pyruvate Under Aerobic Conditions
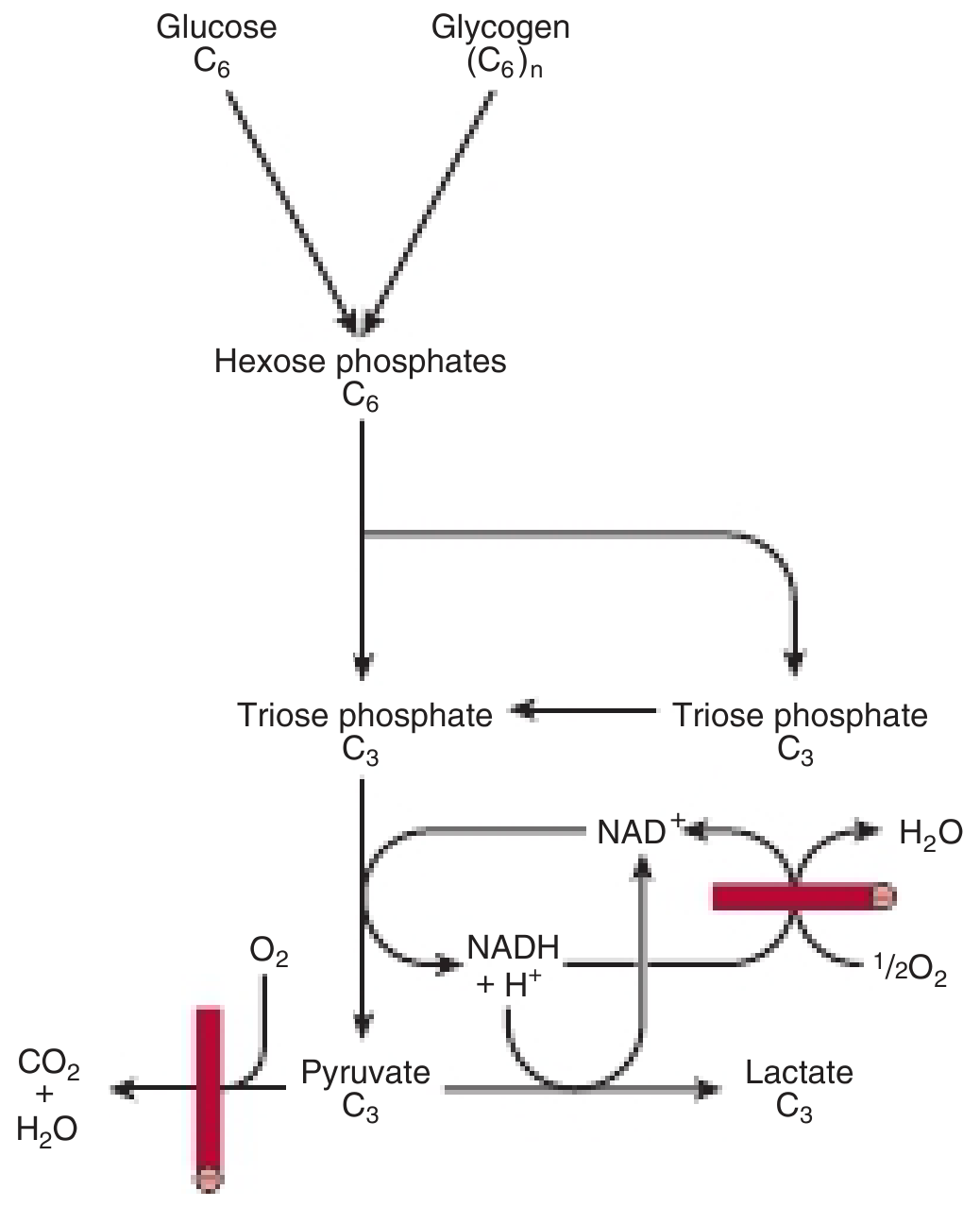
Under aerobic conditions, pyruvate enters the mitochondrial matrix via a proton symporter and undergoes oxidative decarboxylation by the pyruvate dehydrogenase complex (PDC):
Pyruvate + CoA + NAD⁺ → Acetyl-CoA + CO₂ + NADH
Acetyl-CoA then enters the TCA (Krebs) cycle → generating 3 NADH + 1 FADH₂ + 1 GTP per turn (×2 per glucose).
The NADH and FADH₂ are reoxidized by the electron transport chain (ETC) via oxidative phosphorylation, generating the bulk of ATP.
6. Total ATP Yield from Complete Aerobic Oxidation
(Harper's Table 17-1)
| Stage | Reaction | Coenzyme / Product | ATP yield |
|---|---|---|---|
| Glycolysis | G3P dehydrogenase | 2 NADH (cytosolic) | 5 (via malate-aspartate shuttle) |
| Glycolysis | Phosphoglycerate kinase | Substrate-level | 2 |
| Glycolysis | Pyruvate kinase | Substrate-level | 2 |
| Glycolysis | Hexokinase + PFK-1 | ATP consumed | −2 |
| Glycolysis net | 7 | ||
| Pyruvate DH | 2× Pyruvate → Acetyl-CoA | 2 NADH | 5 |
| TCA | Isocitrate DH | 2 NADH | 5 |
| TCA | α-KG DH | 2 NADH | 5 |
| TCA | Succinate thiokinase | 2 GTP | 2 |
| TCA | Succinate DH | 2 FADH₂ | 3 |
| TCA | Malate DH | 2 NADH | 5 |
| TCA net | 25 | ||
| TOTAL | ~30–32 ATP |
Note: If the glycerophosphate shuttle (not malate-aspartate) is used for cytosolic NADH, yield = ~30 ATP. The glycerophosphate shuttle yields only 1.5 ATP/NADH vs 2.5 for malate-aspartate.
7. Regulation of Glycolysis — Three Irreversible Steps
Three enzymes catalyze physiologically irreversible (non-equilibrium) reactions — these are the major regulatory points (Harper's):
① Hexokinase / Glucokinase (Step 1)
- HK: Allosterically inhibited by G6P (product inhibition) → prevents glucose accumulation → found in muscle/brain
- GK (liver/β-cell): High Km, not inhibited by G6P; acts as a "glucose sensor"; induced by insulin; found in the nucleus bound to glucokinase regulatory protein in the fasted state
② Phosphofructokinase-1 — PFK-1 (Step 3) — Rate-limiting step
| Activators | Inhibitors |
|---|---|
| AMP, ADP | ATP (allosteric) |
| Fructose-2,6-bisphosphate (F2,6-BP) — most potent activator | Citrate |
| Pi | Low pH |
- F2,6-BP is the principal regulator in liver: insulin ↑ F2,6-BP (via PFK-2 activation) → stimulates glycolysis; glucagon ↓ F2,6-BP → inhibits glycolysis, promotes gluconeogenesis
③ Pyruvate Kinase (Step 10)
- Inhibited by: ATP, acetyl-CoA, alanine (signals of energy/nutrient sufficiency)
- Activated by: Fructose-1,6-bisphosphate (feedforward activation — when PFK-1 is active, PK is primed)
- Liver PK (L-type): Additionally regulated by hormones via phosphorylation (glucagon → cAMP → PKA → phosphorylates PK → inhibits it; insulin dephosphorylates → activates it)
8. NADH Shuttle Systems (Critical for Aerobic ATP Yield)
NADH cannot cross the inner mitochondrial membrane. Two shuttles transfer its reducing equivalents:
| Shuttle | Tissues | Mitochondrial acceptor | ATP per NADH |
|---|---|---|---|
| Malate-aspartate shuttle | Heart, liver, kidney | NAD⁺ → NADH | 2.5 ATP |
| Glycerophosphate shuttle | Brain, skeletal muscle, white fat | FAD → FADH₂ | 1.5 ATP |
9. Aerobic vs. Anaerobic Glycolysis — Comparison
| Feature | Aerobic Glycolysis | Anaerobic Glycolysis |
|---|---|---|
| O₂ required? | Yes (to reoxidize cytosolic NADH) | No |
| End product | Pyruvate → Acetyl-CoA → TCA | Lactate (via LDH) |
| ATP yield | ~30–32 ATP/glucose | 2 ATP/glucose |
| NADH fate | Enters ETC via shuttles | Reoxidized by pyruvate → lactate |
| Tissues | Most aerobic tissues (heart, liver, brain) | RBC (no mitochondria), renal medulla, WBCs, exercising muscle |
| NAD⁺ regeneration | Via ETC | Via lactate dehydrogenase (LDH) |
10. Clinical Relevance (Harrison's, 22nd Ed)
| Condition | Mechanism | Result |
|---|---|---|
| Lactic acidosis | Hypoxia → O₂ unavailable → anaerobic glycolysis → lactate accumulates, exceeds buffer capacity | ↓ pH, lactic acidemia |
| Warburg effect (cancer) | Tumours preferentially use aerobic glycolysis → lactate even with O₂ ("aerobic glycolysis") | Basis of PET imaging (↑¹⁸F-FDG uptake) |
| Thiamine (B₁) deficiency | Pyruvate dehydrogenase (requires TPP) impaired → pyruvate accumulates → lactic acidosis | Wernicke's encephalopathy, beriberi |
| G6PD deficiency | G6P cannot enter pentose phosphate pathway → oxidative stress in RBCs | Haemolytic anaemia |
| Brain hypoxia | Neurons rely on aerobic glucose oxidation for ATP; O₂ interruption → consciousness lost within 10 seconds | Ischaemic stroke, cardiac arrest |
| Glycogen storage diseases (Types I, III, V, VIII) | Inability to mobilize glucose → impaired glycolysis during fasting/exercise | Fasting hypoglycaemia, exercise intolerance |
| Arsenate poisoning | Arsenate substitutes for Pi in reaction 6 → forms 1-arseno-3-phosphoglycerate (unstable) → bypasses ATP generation at step 7 | Loss of ATP generation from glycolysis |
| Fluoride poisoning | Inhibits enolase (step 9) | Blocks glycolysis |
| 2,3-BPG in RBCs | Bypass of phosphoglycerate kinase step → 2,3-BPG formed → binds Hb → ↓ O₂ affinity | Adaptation to hypoxia; impaired in hexokinase deficiency |
Flowchart Summary
GLUCOSE (C₆)
│ ATP ──── Hexokinase / Glucokinase [Step 1] ← Inhibited by G6P (HK)
▼
Glucose-6-Phosphate
│ ──── Phosphoglucose isomerase [Step 2]
▼
Fructose-6-Phosphate
│ ATP ──── PFK-1 [Step 3] ← RATE-LIMITING; activated by AMP, F2,6-BP
▼ inhibited by ATP, citrate
Fructose-1,6-Bisphosphate
│ ──── Aldolase [Step 4]
├──────────────────────────┐
▼ ▼
Glyceraldehyde-3-P ←────── DHAP [Triose phosphate isomerase, Step 5]
×2 below ↓
│ NAD⁺→NADH ──── G3P Dehydrogenase [Step 6] ← 2 NADH generated
▼
1,3-Bisphosphoglycerate
│ ADP→ATP ──── Phosphoglycerate Kinase [Step 7] ← 2 ATP (substrate-level)
▼
3-Phosphoglycerate
│ ──── Phosphoglycerate mutase [Step 8]
▼
2-Phosphoglycerate
│ ──── Enolase [Step 9] ← Inhibited by fluoride
▼
Phosphoenolpyruvate (PEP)
│ ADP→ATP ──── Pyruvate Kinase [Step 10] ← 2 ATP; activated by F1,6-BP
▼
PYRUVATE (×2)
│
├─ [AEROBIC: O₂ present] ──→ PDC → ACETYL-CoA → TCA cycle → ETC → ~30-32 ATP total
│
└─ [ANAEROBIC: no O₂] ───→ LDH → LACTATE + NAD⁺ (regenerated) → 2 ATP total
Sources: Harper's Illustrated Biochemistry 32nd Ed (Ch. 17); Basic Medical Biochemistry: A Clinical Approach 6th Ed (Ch. 22); Harrison's Principles of Internal Medicine 22nd Ed (2025)
This is a shared conversation. Sign in to Orris to start your own chat.