Explain the mechanism of diabetic nephropathy vs membranous nephropathy vs minimal change disease vs other nephrotic syndromes
Nephrotic Syndromes: Mechanisms Compared
Overview: The Common Final Pathway
1. Diabetic Nephropathy
Core Mechanism
| Pathway | Mechanism | Consequence |
|---|---|---|
| Advanced glycation end-products (AGEs) | Glucose non-enzymatically glycates GBM collagen and mesangial matrix proteins → cross-linking, reduced turnover | GBM thickening, mesangial expansion |
| Polyol pathway | Aldose reductase converts glucose → sorbitol → fructose; depletes NADPH and glutathione | Oxidative stress, endothelial dysfunction |
| PKC activation | Hyperglycemia activates protein kinase C → upregulates TGF-β, VEGF, plasminogen activator inhibitor-1 | Pro-fibrotic signaling, GBM thickening |
| RAAS overactivation | Intraglomerular hypertension → angiotensin II → mesangial expansion, TGF-β → fibrosis | Glomerulosclerosis; RAAS blockers are now standard therapy |
Hemodynamic Component
Genetic Susceptibility
Morphology
- Light microscopy: Diffuse and nodular mesangial expansion; Kimmelstiel-Wilson nodules (pathognomonic); GBM thickening; hyaline arteriolosclerosis of both afferent and efferent arterioles (afferent-only involvement is typical of hypertension)
- Immunofluorescence: Linear IgG and albumin trapping in GBM (non-immune; due to increased permeability and protein insudation — not antibody-mediated)
- EM: GBM thickening, diffuse foot process effacement in advanced disease
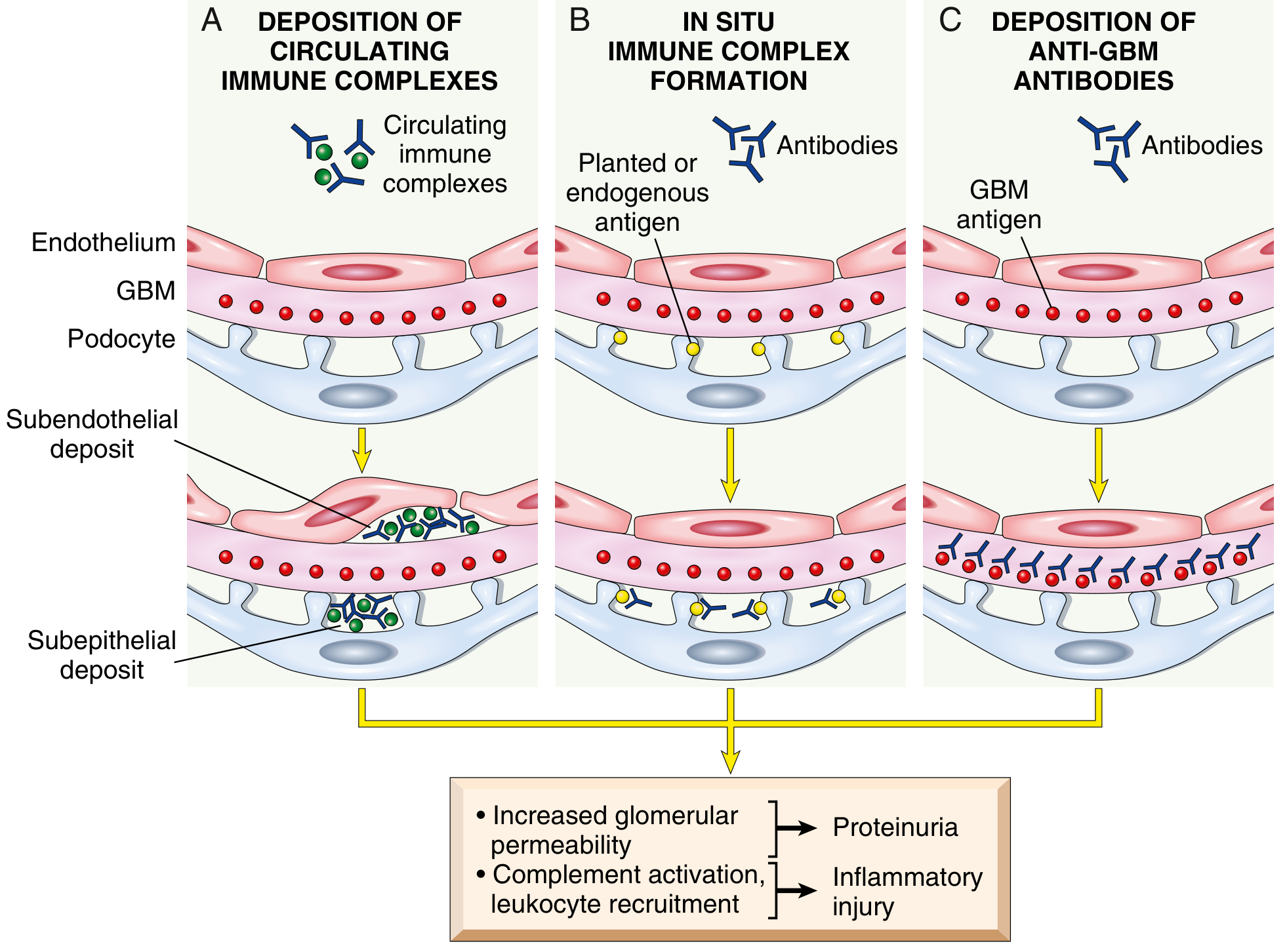
2. Membranous Nephropathy (MN)
Core Mechanism
- IgG4 does not activate the classical complement pathway
- Instead, activates the lectin (MBL) complement pathway → generates membrane attack complex (C5b-9)
- C5b-9 inserts into the podocyte membrane → podocyte activation and injury → disruption of the slit diaphragm and cytoskeleton → proteinuria
- Importantly: little to no inflammation (no leukocyte infiltration), because IgG4 is non-inflammatory
Secondary Causes
- Form circulating immune complexes → trapped in capillary wall → reform subepithelially
- Deposit directly in the subepithelial space where circulating antibody binds them
- Trigger oncogenic virus-related immune dysregulation
Morphology
- Light microscopy: Diffuse GBM thickening; "spike and dome" pattern on silver stain (spikes = projections of new GBM matrix between deposits)
- Immunofluorescence: Granular IgG deposits along GBM in a subepithelial pattern ("lumpy-bumpy")
- EM: Electron-dense subepithelial deposits; foot process effacement
3. Minimal Change Disease (MCD)
Core Mechanism
| Candidate | Evidence |
|---|---|
| T cell-derived permeability factor | T cells are activated in MCD; T cell hybridomas secrete a factor causing heavy proteinuria in rats |
| IL-13 | Overexpressed by T cells in MCD; overexpression in rats causes histologic MCD |
| Angiopoietin-like 4 (ANGPTL4) | Overexpressed by podocytes in MCD; causes proteinuric response; reducible by steroids and N-acetyl-D-mannosamine |
| CD80 (B7.1) | Elevated in urine and podocytes; correlates with disease activity; normally expressed only on dendritic cells/B cells |
| Anti-nephrin antibodies | A subset of MCD patients have IgG antibodies against nephrin (slit diaphragm protein); recently recognized — Robbins, Cotran & Kumar |
Morphology
- Light microscopy: Completely normal — hence "minimal change"
- Immunofluorescence: Negative — no immune deposits
- EM: Diffuse effacement (fusion) of podocyte foot processes ± vacuolization, microvillus formation — the only diagnostic abnormality
- Proximal tubular cells laden with protein droplets and lipid (reabsorption artifacts)
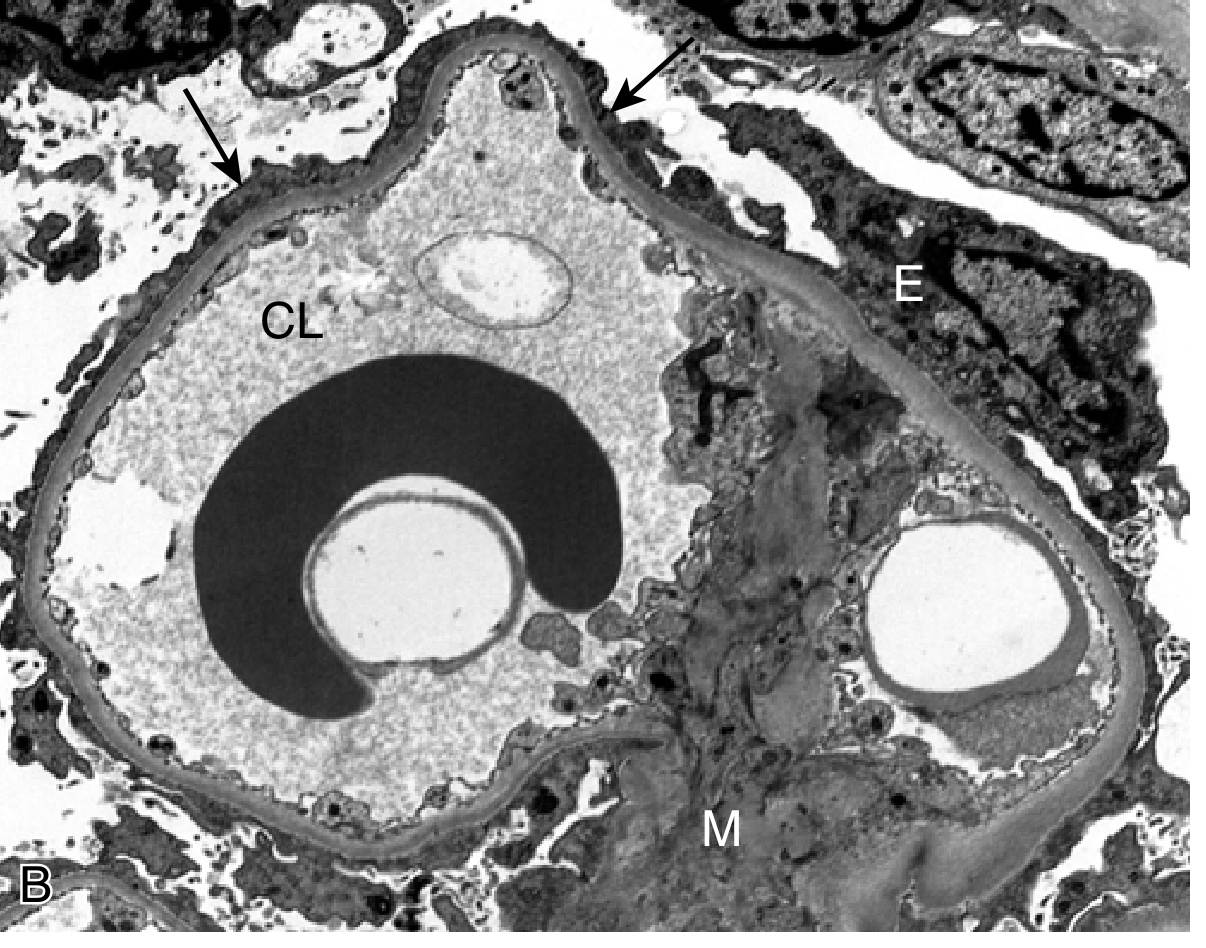
4. Focal Segmental Glomerulosclerosis (FSGS)
Core Mechanism
- Nephron loss → glomerular hypertension (maladaptive hyperfiltration)
- Obesity (same mechanism — hyperfiltration)
- HIV (collapsing variant — HIVAN) and COVID-19 (COVAN) — especially in APOL1 high-risk individuals
- Heroin and some therapeutic drugs
Collapsing FSGS Variant
Morphology
- Light microscopy: Focal (some glomeruli) and segmental (part of tuft) obliteration of capillary loops; mesangial matrix increase; hyalinosis (IgM and protein insudation); foamy macrophages
- Immunofluorescence: Non-specific IgM and C3 trapping in sclerotic areas
- EM: Diffuse foot process effacement (in primary, acute-phase FSGS); focal podocyte detachment with GBM denudation
5. Membranoproliferative GN (MPGN)
Mechanism
6. C3 Glomerulopathy (Dense Deposit Disease / C3GN)
Mechanism
- C3 nephritic factor (C3NeF): autoantibody that binds and stabilizes C3bBb (the alternative pathway C3 convertase) → persistent C3 activation → hypocomplementemia (low C3; normal C1, C4)
- Factor H mutations: loss of the main inhibitor of the alternative pathway
7. Amyloidosis
Mechanism
Summary Comparison Table
| Feature | Diabetic Nephropathy | Membranous Nephropathy | Minimal Change Disease | FSGS |
|---|---|---|---|---|
| Primary mechanism | Metabolic/hemodynamic (AGEs, PKC, RAAS) | Anti-PLA2R IgG4 → subepithelial immune deposits → C5b-9 | Circulating factor (IL-13, ANGPTL4, anti-nephrin?) | Circulating permeability factor / genetic podocyte mutation |
| Podocyte injury | Indirect (metabolic stress, glomerular hypertension) | C5b-9 insertion into podocyte | Direct (unknown factor) | Direct loss → denuded GBM |
| Light microscopy | Kimmelstiel-Wilson nodules, GBM thickening | GBM thickening, "spikes" | Normal | Focal, segmental sclerosis |
| Immunofluorescence | Linear IgG/albumin (non-specific trapping) | Granular subepithelial IgG | Negative | Non-specific IgM/C3 in sclerotic areas |
| EM | GBM thickening, foot process effacement (late) | Subepithelial electron-dense deposits | Diffuse foot process effacement only | Foot process effacement + podocyte detachment |
| Proteinuria type | Non-selective (advanced disease) | Non-selective | Selective (albuminuria) | Non-selective |
| Steroid response | None | Variable/resistant (anti-CD20 preferred for primary MN) | Excellent (>90% children) | Generally poor |
| Complement | Normal | Normal | Normal | Normal |
| Key biomarker | HbA1c; GFR trajectory | Anti-PLA2R antibody titer | — | APOL1 genotype (risk) |
Convert each and everything to diagram and sketch
Medical education diagram titled "Nephrotic Syndrome — Common Final Pathway". Clean anatomical sketch style with labels. Shows a cross-section of a glomerular capillary wall with three layers labeled: Endothelium (bottom), Glomerular Basement Membrane (GBM) (middle), Podocyte with foot processes (top). Arrows from multiple disease boxes on the left (Diabetic Nephropathy, Membranous Nephropathy, Minimal Change Disease, FSGS, MPGN, Amyloidosis) all converge with arrows pointing to "Podocyte Injury / Foot Process Effacement" at the top of the capillary wall. From the podocyte injury, a large bold arrow points right to a box showing the nephrotic syndrome consequences: Proteinuria >3.5g/day → Hypoalbuminemia → Generalized Edema; Hyperlipidemia & Lipiduria. Bottom note: "All nephrotic syndromes share the same final pathway — the mechanisms that injure podocytes differ." Beige and white background, blue and navy labels, clear educational medical illustration style.
Medical education mechanism diagram titled "Diabetic Nephropathy — Pathogenesis". Sketch/diagram style, clean educational illustration. Central starting point: "Chronic Hyperglycemia" in a large box at the top. Four arrows branch downward to four parallel pathway boxes: 1) "AGE Formation — Non-enzymatic glycation of GBM collagen → cross-linking → GBM thickening, mesangial expansion"; 2) "Polyol Pathway — Aldose reductase: Glucose → Sorbitol → depletes NADPH/glutathione → Oxidative Stress → endothelial dysfunction"; 3) "PKC Activation — Protein Kinase C activated → upregulates TGF-β, VEGF, PAI-1 → Pro-fibrotic signaling → GBM thickening"; 4) "RAAS Overactivation — Intraglomerular hypertension → Angiotensin II → Mesangial expansion → Glomerulosclerosis". All four boxes have arrows pointing down to "Podocyte Injury + Glomerulosclerosis". Below that: "Kimmelstiel-Wilson nodules (LM) | GBM thickening | Foot process effacement (late, EM) | Linear IgG/albumin (non-specific, IF)". Left side inset: glomerular cross-section sketch showing afferent arteriolar vasodilation → hyperfiltration → mechanical stress on podocyte. White background, dark navy text, color-coded pathway boxes.
Medical education mechanism diagram titled "Membranous Nephropathy — Pathogenesis". Clean anatomical sketch/diagram style. Top section: "Idiopathic (70%): Anti-PLA2R IgG4 autoantibodies" with an antibody icon. Show a detailed cross-section of the glomerular capillary wall: Endothelium at bottom, GBM in middle, Podocyte with foot processes at top. Between the GBM outer surface and the podocyte foot process base: electron-dense "Subepithelial Immune Deposits" shown as bumps/dots labeled. Arrow from deposits to: "IgG4 — does NOT activate classical complement". Then arrow to: "Activates MBL/Lectin Complement Pathway → generates C5b-9 (Membrane Attack Complex) → inserts into podocyte membrane → podocyte activation/injury → slit diaphragm disruption → proteinuria". Right side inset box: "Secondary MN causes: Cancer (lung, gastric, prostate) | HBV, HCV | Drugs (NSAIDs, penicillamine) | SLE". Bottom findings row: "LM: GBM thickening, spike-and-dome | IF: Granular IgG subepithelial (lumpy-bumpy) | EM: Subepithelial electron-dense deposits". White background, teal/navy color scheme, educational medical style.
Medical education mechanism diagram titled "Minimal Change Disease — Pathogenesis". Clean sketch/diagram educational style. Shows a podocyte cell prominently in the center with its foot processes projecting down onto the GBM. Multiple circulating factors shown as labeled arrows arriving at the podocyte from the blood: "IL-13 (from activated T cells)", "Angiopoietin-like 4 (ANGPTL4) overexpressed by podocyte", "CD80/B7.1 expressed on podocyte (correlates with disease activity)", "Anti-nephrin IgG antibodies (subset of cases)". Large arrow from "Circulating Factor (T cell / B cell origin — exact molecule unknown)" pointing to podocyte. Result: "Foot Process Effacement (diffuse)" shown as flattened/fused foot processes. Right side: "Steroids ↓ ANGPTL4, reverse effacement → remission of proteinuria". Bottom findings row: "LM: NORMAL glomerulus | IF: NEGATIVE — no deposits | EM: Diffuse foot process effacement ONLY | Proteinuria: SELECTIVE (mainly albumin)". Top right badge: "Most common nephrotic syndrome in children | >90% respond to steroids". White background, warm orange and navy educational diagram style.