Explain the mechanism of acute respiratory distress syndrome
Reading File
Finding Sources
Searching PubMed
"acute respiratory distress syndrome"[MeSH Terms] AND pathophysiology
Reading File
Reading File
Searching Images
ARDS acute respiratory distress syndrome pathophysiology mechanism diagram
Reading File
Finding Sources
Reading File
Reading File
Reading File
Finding Sources
Reading File
Reading File
Loading Image
Loading Image
I now have all the material needed. Let me compose a comprehensive, well-structured answer.
Acute Respiratory Distress Syndrome (ARDS): Mechanism
Definition and Berlin Criteria
ARDS is a syndrome of acute hypoxemic respiratory failure caused by diffuse inflammatory lung injury. The 2012 Berlin Definition requires:
- Bilateral pulmonary infiltrates on chest imaging
- PaO₂/FiO₂ (P/F) ratio <300 mmHg on ≥5 cmH₂O PEEP
- Onset within 7 days of a recognized insult
- Respiratory failure not fully explained by cardiac failure or fluid overload
Severity is classified as mild (P/F 200–300), moderate (P/F 100–200), and severe (P/F <100).
Core Pathophysiology: Breakdown of the Alveolar-Capillary Barrier
The unifying mechanism is increased alveolar-capillary permeability — not elevated hydrostatic pressure (as in cardiogenic pulmonary edema). This leads to protein-rich exudative fluid flooding the alveoli.
The alveolar-capillary unit has two barriers:
- Microvascular endothelium — normally the primary barrier to fluid; disrupted early in ARDS
- Alveolar epithelium — normally even tighter; injury here is particularly damaging because type II pneumocytes produce surfactant and drive alveolar fluid clearance via active Na⁺ transport
When both barriers are disrupted, protein-rich fluid, plasma proteins, and inflammatory cells pour into the interstitium and alveoli.
Phases of ARDS: Diffuse Alveolar Damage (DAD)

Figure: Time course of ARDS. Exudative phase features alveolar edema and hyaline membrane formation (days 0–7); proliferative phase involves interstitial inflammation and early fibrosis (days 7–21); fibrotic phase occurs in a subset of patients with persistent disease. — Harrison's Principles of Internal Medicine, 22nd ed.
1. Exudative Phase (Days 0–7)
- Endothelial and type I pneumocyte injury → loss of tight junctions → protein-rich exudate floods alveoli
- Proinflammatory cytokines (IL-1β, IL-6, IL-8, TNF-α) and lipid mediators (leukotriene B₂) are elevated
- Massive neutrophil recruitment into pulmonary vasculature and alveoli (see below)
- Condensed plasma proteins + cellular debris + dysfunctional surfactant form hyaline membranes
- Pulmonary vascular injury: microthrombi, fibrocellular proliferation, obliteration of small vessels
- Result: profound hypoxemia (right-to-left shunting from collapsed/fluid-filled alveoli) + dead-space ventilation + reduced lung compliance
2. Proliferative Phase (Days 7–21)
- Hyaline membranes are reorganized; type II pneumocytes proliferate to replace lost type I cells
- Interstitial inflammation remains prominent
- Early fibrosis appears with collagen deposition (N-terminal procollagen peptide III detectable in BAL fluid within 24 hours of onset — fibroproliferation may begin simultaneously with inflammation)
- Pulmonary capillary obliteration and interstitial/alveolar collagen deposition
- Neutrophil numbers decline; macrophages become dominant
3. Fibrotic Phase (>21 days, subset of patients)
- Pulmonary fibrosis, bullae formation, and cyst formation in those with persistent ARDS
- Associated with prolonged mechanical ventilation and poor outcomes
The Neutrophil: Central Effector of Lung Injury
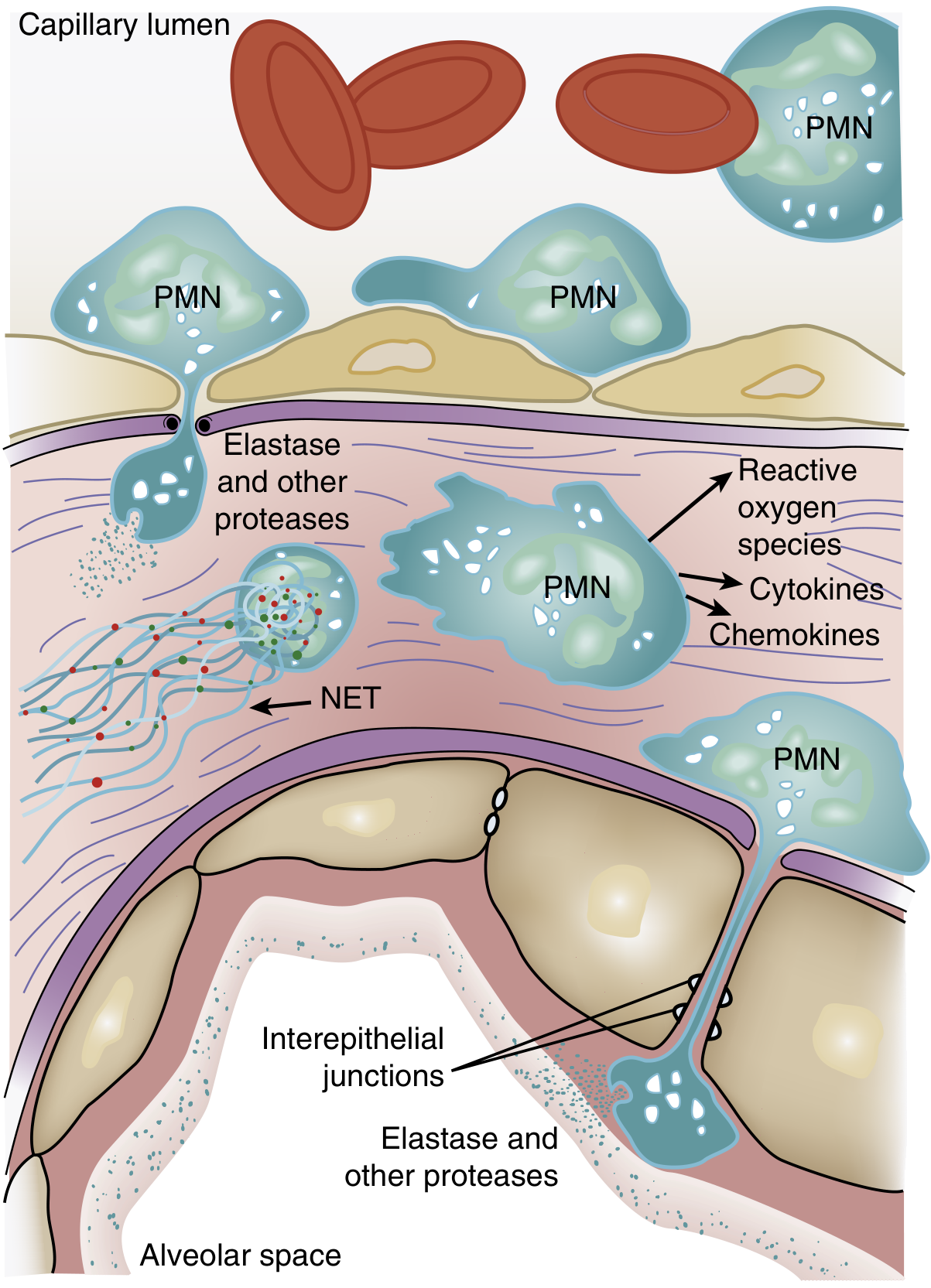
Figure: Activated PMNs exit the capillary lumen, transmigrate across the alveolar-capillary membrane, and release cytotoxic compounds. — Murray & Nadel's Textbook of Respiratory Medicine
One of the earliest findings in ARDS is transient leukopenia — reflecting pulmonary neutrophil sequestration before hypoxemia even develops. This occurs because:
- Pulmonary capillaries are narrower than neutrophil diameter — cells must deform to pass through
- Activated neutrophils become "stiff" (actin cytoskeleton change) and cannot deform → sequestration in capillaries
- Sequestered PMNs induce endothelial barrier breakdown, enhancing further transmigration into the interstitium
Neutrophil Weapons that Cause Tissue Damage
| Mediator | Mechanism of Injury |
|---|---|
| Reactive oxygen species (ROS) | Oxidative damage to endothelial and epithelial membranes |
| Neutrophil elastase (NE) | Degrades cadherins (adherens junctions) → alveolar flooding; degrades growth factors and cytokines |
| Metalloproteinases | Extracellular matrix destruction |
| Defensins / cationic peptides | Direct membrane disruption |
| TNF-α, IL-1β | Amplify inflammatory cascade |
| Neutrophil Extracellular Traps (NETs) | DNA/histone/antimicrobial webs → endothelial damage + thrombus formation |
NETs are particularly important: in sepsis, massive NET release causes NET-associated endothelial damage and microvascular thrombosis. Mouse models show NET formation in lungs is accompanied by severe structural destruction.
Signal Transduction Amplification
- p38 MAPK (activated by LPS) → stimulates TNF-α production and macrophage inflammatory protein-2 (MIP-2) → further neutrophil chemotaxis
- PI3K-γ (activated by IL-8 and bacterial peptides in neutrophils) → promotes neutrophil accumulation and cytokine production
Surfactant Dysfunction
- Surfactant proteins (SP-A, SP-B, SP-C, SP-D) are reduced and dysfunctional in ARDS
- Phospholipase A₂ (released in pancreatitis) enzymatically degrades surfactant → alveolar collapse
- Neutrophil elastase and plasma proteins entering alveoli directly damage surfactant lipids and proteins
- Loss of surfactant → increased alveolar surface tension → tendency to collapse (atelectasis) and ventilation-perfusion mismatch
Pulmonary Vascular Consequences
- Hypoxic vasoconstriction in poorly ventilated units
- Microvascular thrombosis from NETs, activated platelets, and fibrin deposition
- Compression of vessels by positive-pressure ventilation
- Net result: pulmonary hypertension with increased dead space → hypercapnia compounds hypoxemia
Physiological Consequences
| Abnormality | Mechanism |
|---|---|
| Severe hypoxemia | Intrapulmonary shunt (flooded/atelectatic alveoli) |
| Hypercapnia | Increased dead space (obliterated/compressed vessels) |
| Reduced compliance | Alveolar edema, atelectasis, loss of surfactant |
| Pulmonary hypertension | Hypoxic vasoconstriction, microthrombosis |
| Respiratory muscle fatigue | Increased work of breathing against stiff lungs |
Common Precipitants
ARDS can arise from direct (pulmonary) or indirect (extrapulmonary) lung injury:
- Direct: pneumonia (most common), aspiration, pulmonary contusion, inhalation injury
- Indirect: sepsis (most common overall), severe pancreatitis, trauma, transfusion-related (TRALI), burns
In pancreatitis, for example, elastase and lipase released systemically increase vascular permeability, phospholipase A₂ degrades surfactant, and TNF-α/IL-8 drive neutrophil recruitment to the lung — the same final common pathway regardless of the inciting cause.
Sources: Murray & Nadel's Textbook of Respiratory Medicine (block31); Harrison's Principles of Internal Medicine, 22nd ed. (block32); Goldman-Cecil Medicine (block12); Sabiston Textbook of Surgery (block11)
This is a shared conversation. Sign in to Orris to start your own chat.