Propofol history, moa, everything
Reading File
Finding Sources
Finding Sources
Searching PubMed
propofol pharmacology mechanism action
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Reading File
Reading File
I now have comprehensive information to write an authoritative, thorough answer on propofol. Let me compile the full response.
Propofol: Complete Overview
Chemical Structure
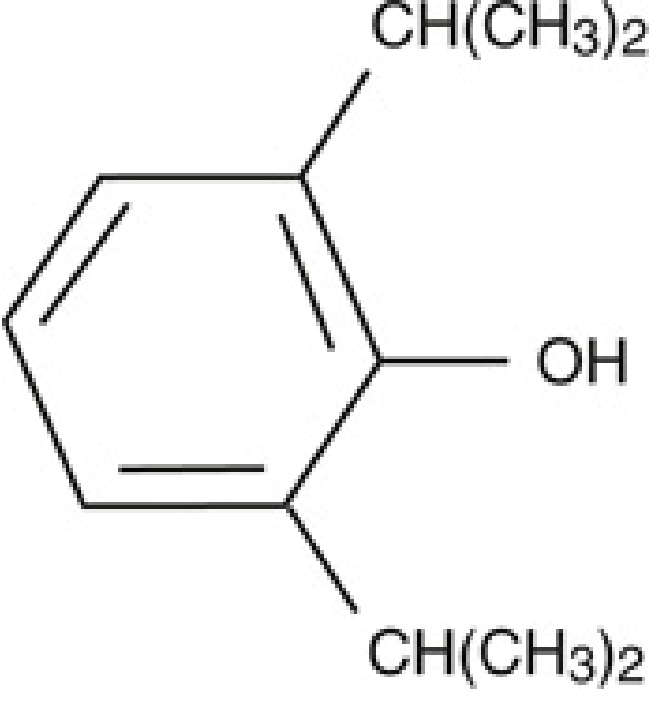
Propofol (2,6-diisopropylphenol) is a short-chain alkylphenol. It has a benzene ring with a hydroxyl group flanked symmetrically by two isopropyl substituents at the 2 and 6 positions. This structure is highly lipid-soluble and virtually insoluble in water - which dictated its entire formulation history.
History
- 1970s (UK): Propofol was developed by Imperial Chemical Industries (ICI) under the compound name ICI 35868, building on research into the sedative properties of phenol derivatives in mice.
- 1977: First released clinically, formulated in Cremophor EL (polyoxylated castor oil). It was rapidly withdrawn after reports of serious anaphylactic reactions, attributable to the Cremophor vehicle.
- 1986: Reintroduced in a reformulated lipid emulsion - soybean oil/propofol/water - which resolved the anaphylaxis problem. This formulation remains in use today.
- 1990s: EDTA (ethylenediaminetetraacetic acid) was added to the emulsion as a bacteriostatic agent to deter microbial growth in the high-fat emulsion.
- Post-1986: Propofol rapidly displaced barbiturates (primarily thiopental) as the dominant IV induction agent. Today it is the most commonly used IV anesthetic in the world.
- 2008: The FDA approved fospropofol disodium (Lusedra), a water-soluble prodrug of propofol, for monitored anesthesia care in adults.
Formulation
Current standard formulation (1%):
- 1% propofol (10 mg/mL)
- 10% soybean oil (lipid carrier)
- 1.2% purified egg phospholipid (emulsifier)
- 2.25% glycerol (tonicity adjustment)
- Sodium hydroxide (pH adjustment to ~7)
- EDTA (bacteriostatic)
The result is the characteristic milky white, slightly viscous appearance. It is stable at room temperature, not light-sensitive, and can be diluted with 5% dextrose in water. A 2% formulation is available in Europe. The lipid emulsion means propofol is calorically significant (~1.1 kcal/mL) - relevant in ICU patients on prolonged infusions.
Mechanism of Action
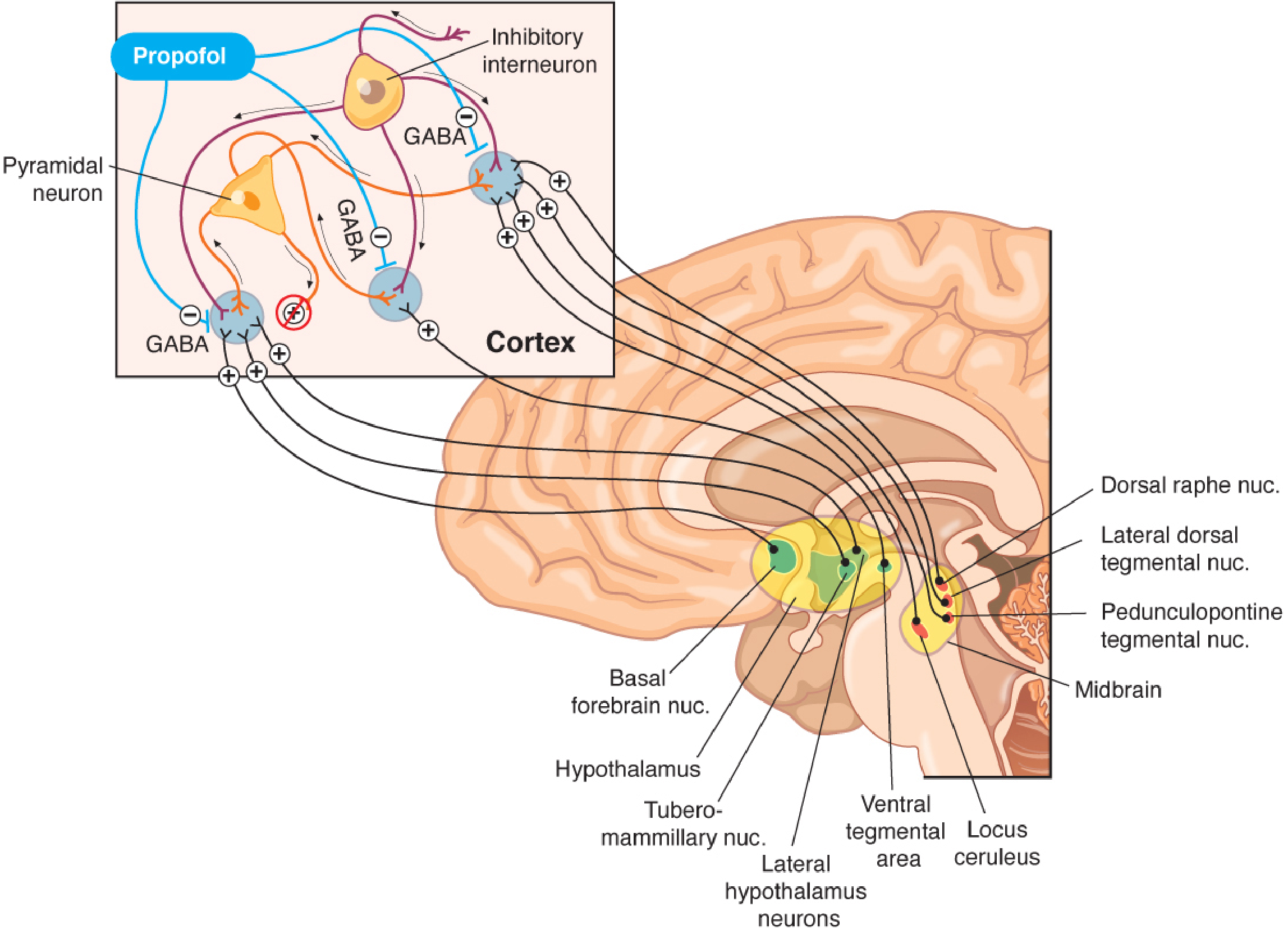
The primary mechanism is potentiation of GABA-A receptor-mediated chloride currents.
GABA-A Receptor Modulation
- Propofol binds to specific sites on the GABA-A receptor (a ligand-gated chloride channel), distinct from the benzodiazepine binding site.
- At clinically relevant concentrations, it enhances (potentiates) the effect of endogenous GABA - increasing the frequency and duration of chloride channel opening, causing neuronal hyperpolarization and reduced excitability.
- At higher (supratherapeutic) concentrations, propofol can directly activate GABA-A receptor channels even in the absence of GABA.
- Structural studies confirm binding sites for propofol on GABA-A receptors; propofol appears to act preferentially at receptors containing α2 and α3 subunits.
Where in the Brain?
As shown in the diagram above, propofol acts at multiple levels of the ascending arousal system:
- GABAergic inhibitory interneurons suppress pyramidal cortical neurons
- Arousal inputs from thalamus, hypothalamus (tuberomammillary nucleus), locus ceruleus, dorsal raphe nucleus, basal forebrain, and midbrain are all dampened
- The net result is counteraction of excitatory ascending inputs, producing unconsciousness
Other Receptor Interactions
Propofol's MOA is not exclusively GABA-A:
- Inhibition of NMDA glutamate receptors (contributes to amnesia, analgesia at high doses)
- Modulation of glycine receptors (inhibitory)
- Inhibition of sodium channels
- Activation of K+ ATP channels (contributes to vasodilation)
- Inhibition of the hyperpolarization-activated cyclic nucleotide-gated (HCN) channels
- Antioxidant effects (free radical scavenging) independent of receptor activity
The exact mechanism of the transition from consciousness to unconsciousness is not fully elucidated - the "unitary theory" of anesthesia is now considered incorrect.
Pharmacokinetics
Multicompartment Model
Propofol follows a three-compartment pharmacokinetic model:
| Parameter | Value |
|---|---|
| Initial distribution half-life | 1-8 minutes |
| Secondary (slow) distribution half-life | 30-70 minutes |
| Elimination half-life | 2-24 hours |
| Volume of distribution (steady-state) | ~60 L/kg (very large - due to high lipophilicity) |
| Plasma protein binding | ~98% (mainly albumin) |
| Clearance | 20-30 mL/kg/min (~1.5 L/min) - exceeds hepatic blood flow |
Metabolism
- Primarily hepatic - conjugation to glucuronide and sulfate metabolites, which are water-soluble and renally excreted. Less than 3% excreted as unchanged propofol.
- Because clearance exceeds hepatic blood flow (normal ~15 mL/kg/min), significant extrahepatic metabolism occurs - kidneys and lungs each account for up to 30% of clearance.
- This explains why hepatic or renal disease does not significantly alter pharmacokinetics - no dose adjustment is required.
Context-Sensitive Half-Time (CSHT)
A key clinical advantage:
- Infusion <3 hours: CSHT ~10 minutes
- Infusion up to 8 hours: CSHT <40 minutes
- This predictable, short CSHT allows reliable titration and rapid emergence even after prolonged infusions.
Special Populations
| Population | Effect |
|---|---|
| Elderly (>80 years) | Need ~50% less dose than 20-year-olds; reduced cardiac output and clearance |
| Children >3 years | Larger central compartment (+50%), faster clearance (+25%) - need more mg/kg |
| Children <3 years | Even larger central compartment and clearance |
| Hepatic disease | Larger Vd, slightly prolonged elimination half-life; no significant dose adjustment needed |
| Hemorrhagic shock | Slower clearances, 2.7x increased sensitivity - shift concentration-effect curve leftward |
| Obesity | Use lean body weight for dosing |
Drug Interactions
- Midazolam + propofol: midazolam raises propofol plasma levels by ~25% (reduces propofol clearance)
- Alfentanil/fentanyl/remifentanil: opioids reduce propofol clearance and distribution; propofol concentrations increase 20-30% when combined with midazolam + alfentanil
- Epidural blockade (e.g., 20-segment ropivacaine): reduces propofol requirement by ~30% (likely via reduced hepatic/renal blood flow)
Pharmacodynamics
Dose-Response Spectrum
| Plasma level (approx.) | Clinical effect |
|---|---|
| Sub-sedative | Antiemetic |
| Low sedative | Anxiolysis, sedation |
| Higher sedative | Paradoxical excitation (disinhibition, unpredictable movement) |
| ~3 mcg/mL | Loss of consciousness |
| ~8 mcg/mL | EEG burst suppression |
| Higher | Isoelectric EEG |
CNS Effects
- EEG pattern: At low doses: increased beta activity. At induction doses: deep non-REM sleep pattern (increased low-frequency, high-amplitude; increased alpha and delta, decreased beta). Burst suppression at ~8 mcg/mL. Isoelectric at higher doses.
- Neuroprotection:
- Decreases CMRO2 (cerebral metabolic oxygen consumption)
- Decreases ICP (by reducing CBF)
- Note: CPP may also fall (vasodilation reduces MAP), so benefit is not absolute
- Free radical scavenger/antioxidant properties
- Attenuates excitotoxic glutamate pathways
- Anti-inflammatory (decreases TNF-alpha)
- Anticonvulsant: At burst-suppression doses; used to treat status epilepticus. Shortens seizure duration - therefore NOT the preferred agent for ECT induction. Contradictory reports of proconvulsant effects at low doses exist.
- Loss of consciousness reversal: Can be partially reversed by physostigmine (central cholinomimetic), suggesting cholinergic arousal pathways are involved.
- Abuse potential: Emergence from propofol is associated with feelings of well-being or euphoria in some patients. Highest abuse rates among anesthesia providers with easy access. In the US, 18% of academic institutions have reported propofol abuse/diversion in the past decade, with significant mortality among residents. Only fospropofol (not propofol itself) is currently a DEA-scheduled substance.
Cardiovascular Effects
Propofol causes significant, dose-dependent hemodynamic depression - more pronounced after IV bolus than during infusion:
- Systolic blood pressure drops 25-40% after induction dose (2-2.5 mg/kg)
- Mechanisms:
- Decreased systemic vascular resistance (SVR) - 15-25% reduction (vasodilation)
- Decreased cardiac output/index (~15%)
- Decreased stroke volume index (~20%)
- Decreased left ventricular stroke work index (~30%)
- Suppression of sympathetic nervous system tone
- Direct inhibition of baroreceptor response (prevents reflex tachycardia - heart rate does not increase despite hypotension)
- Mechanisms of vasodilation: Direct effect on intracellular smooth muscle calcium, inhibition of prostacyclin synthesis, reduction of angiotensin II-mediated calcium entry, K+ ATP channel activation, nitric oxide stimulation.
- Heart rate: Minimally affected. Baroreflex is inhibited. Propofol decreases cardiac parasympathetic tone dose-dependently. Attenuates heart rate response to atropine.
- Important timing: The hemodynamic effect-site equilibration half-life (~7 min) lags behind the hypnotic effect (~2-3 min) - blood pressure continues to fall for minutes after consciousness is lost.
- Supraventricular tachycardias are suppressed - propofol should be avoided during electrophysiologic studies.
- Myocardial oxygen balance: Myocardial blood flow and oxygen consumption both decrease; global supply-demand ratio likely preserved. However, volatile anesthetics (sevoflurane, desflurane) may be superior in cardiac surgery (lower troponins, better hemodynamic outcomes).
Respiratory Effects
- Respiratory depression is dose-dependent
- Apnea is common with induction doses (brief)
- Maintenance infusion causes decreased tidal volume, increased respiratory rate
- Blunted response to both hypoxia (direct chemoreceptor effect) and hypercarbia
- Potent bronchodilator - direct effect on intracellular calcium homeostasis
Clinical Uses & Dosing
| Indication | Typical Dose |
|---|---|
| Induction (healthy adult) | 1-2.5 mg/kg IV (target blood conc. ~3 mcg/mL) |
| Maintenance (TIVA) | 100-200 mcg/kg/min infusion |
| Conscious sedation | 25-75 mcg/kg/min (lower end) |
| ICU sedation | 5-50 mcg/kg/min |
| Antiemetic (PONV) | Sub-sedative doses (10-20 mg IV bolus) |
Adjust for: Elderly (50% reduction), children (increase per kg), pre-medications, co-administered opioids/benzodiazepines.
Key advantages driving its widespread use:
- Rapid onset AND offset (predictable CSHT)
- Smooth induction and emergence
- PONV prevention - unique antiemetic property even at sub-sedative concentrations (acts at CTZ)
- No contraindication in malignant hyperthermia (unlike volatile agents)
- Suitable for TIVA, neuroanesthesia (burst suppression), cardiac surgery, procedural sedation, ICU
- Can be used for status epilepticus (via burst suppression)
Propofol Infusion Syndrome (PRIS)
PRIS is a rare but potentially fatal complication associated with high-dose, prolonged propofol infusions (typically >4-5 mg/kg/hr for >48 hours, as seen in ICU sedation). Originally described in children, also occurs in adults.
Features (mnemonic: BRAHMA):
- Bradycardia (and other arrhythmias) - ECG may show Brugada-type pattern
- Rhabdomyolysis
- Acidosis (severe metabolic, lactic)
- Hepatomegaly / hepatic failure
- Metabolic (hypertriglyceridemia, lipemia)
- Acute kidney injury / multi-organ failure
Pathophysiology: Impairment of mitochondrial respiratory chain (inhibits Complex I and II), impaired fatty acid beta-oxidation, uncoupling of oxidative phosphorylation.
Management: Stop propofol immediately, hemodialysis/hemofiltration for acidosis, treat rhabdomyolysis, cardiovascular support. The 10% lipid emulsion itself contributes to hypertriglyceridemia and caloric overload.
Adverse Effects Summary
| Adverse Effect | Details |
|---|---|
| Pain on injection | Very common, especially in small veins; using antecubital fossa, pre-treating with lidocaine, or mixing with lidocaine reduces incidence |
| Hypotension | Most clinically significant effect; dose and rate dependent |
| Apnea | Brief, with induction doses |
| PRIS | See above - high-dose, prolonged infusion |
| Hyperlipidemia | From lipid vehicle; monitor TG in prolonged ICU use |
| Bacterial contamination | Fat emulsion is excellent bacterial growth medium; strict aseptic technique; use within 6-12 hours |
| Egg/soy allergy | Theoretical concern; current evidence suggests it is very rarely clinically significant, but caution is advised |
| Urine discoloration | Green/pink urine - benign, from metabolites |
| Abuse potential | Euphoria on emergence; serious risk in healthcare workers |
| Excitatory phenomena | Paradoxical excitation at intermediate doses; occasional myoclonus at induction |
Contraindications / Cautions
- Known allergy to propofol or its emulsion components (rare true contraindication)
- Use caution in egg phospholipid or soy allergy (consult allergy history)
- Hemodynamically unstable patients (hypovolemia, shock, cardiac compromise) - exaggerated hypotension
- Mitochondrial disease - increased PRIS risk
- Hypertriglyceridemia - use caution in prolonged ICU sedation
- Avoid in electrophysiology studies (suppresses arrhythmias, obscures findings)
- Avoid as primary ECT induction agent (shortens seizure duration)
Fospropofol (Lusedra)
A water-soluble prodrug of propofol (FDA-approved 2008 for monitored anesthesia care). Advantages: no pain on injection, no lipid emulsion. Disadvantage: perineal paresthesias and pruritus are common. Currently the only propofol-related substance on the DEA schedule list.
Sources:
- Miller's Anesthesia, 10th Edition, Chapter 21 (IV Anesthetics - Propofol section)
- Barash, Cullen, and Stoelting's Clinical Anesthesia, 9th Edition, Chapter 19 (IV Anesthetics)
- Katzung's Basic and Clinical Pharmacology, 16th Edition, Chapter on GABA-A receptor pharmacology
This is a shared conversation. Sign in to Orris to start your own chat.