Neoplasia notes from robiins pathology
Reading File
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Here are comprehensive Neoplasia notes from Robbins & Kumar Basic Pathology (Robbins Pathology):
NEOPLASIA - Robbins Pathology Notes
1. DEFINITIONS
- Neoplasia = "new growth"; neoplastic cells replicate incessantly due to resistance to normal regulatory influences
- Tumor / Neoplasm = abnormal mass of tissue whose growth exceeds and is uncoordinated with that of normal tissue and persists after the stimulus ceases
- Oncology = study of tumors (from Greek oncos)
- All neoplasms have two basic components:
- Parenchyma - the neoplastic/transformed cells; determines biologic behavior and naming
- Stroma - host-derived supporting connective tissue, inflammatory cells, and blood vessels; provides blood supply; essential for tumor growth; carries on two-way conversation with tumor cells
2. NOMENCLATURE
Benign Tumors
| Origin | Suffix/Name | Example |
|---|---|---|
| Mesenchymal cells | -oma | Fibroma, chondroma, lipoma |
| Epithelial (glandular / solid) | Adenoma | Hepatic adenoma, renal tubular adenoma |
| Epithelial (finger-like projections) | Papilloma | Squamous papilloma |
| Epithelial (projecting into lumen) | Polyp | Colorectal polyp |
| Epithelial (hollow cystic) | Cystadenoma | Ovarian cystadenoma |
Malignant Tumors
| Origin | Name |
|---|---|
| Epithelial tissue | Carcinoma |
| Squamous epithelium | Squamous cell carcinoma |
| Glandular epithelium | Adenocarcinoma |
| Mesenchymal / connective tissue | Sarcoma |
| Fibroblasts | Fibrosarcoma |
| Smooth muscle | Leiomyosarcoma |
| Fat | Liposarcoma |
| Lymphocytes | Lymphoma |
| Hematopoietic cells | Leukemia |
| Both epithelial + mesenchymal | Carcinosarcoma |
Note: Some names break the rule - lymphoma, leukemia, and melanoma are all malignant despite not having "-carcinoma" or "-sarcoma."
Teratoma = tumor containing cells from all three germ layers (ectoderm, mesoderm, endoderm); can be benign or malignant.
3. CHARACTERISTICS OF BENIGN VS. MALIGNANT NEOPLASMS
3.1 Differentiation and Anaplasia
| Feature | Benign | Malignant |
|---|---|---|
| Differentiation | Well-differentiated; resembles tissue of origin | Variable; may be poorly or undifferentiated (anaplastic) |
| Cell morphology | Normal size/shape | Pleomorphism - variation in size/shape |
| Nuclei | Normal | Hyperchromatic, enlarged, irregular; high N:C ratio |
| Mitoses | Rare, normal | Frequent, atypical (tripolar, quadripolar) |
| Architecture | Organized | Disorganized |
| Tumor giant cells | Absent | May be present |
Anaplasia = loss of structural and functional differentiation; hallmark of malignancy. Features:
- Pleomorphism (variation in cell and nuclear size/shape)
- Abnormal nuclear morphology (hyperchromatic, coarse chromatin, prominent nucleoli)
- Atypical mitoses (abnormal spindle formation)
- Loss of polarity
- Tumor giant cells
- Areas of ischemic necrosis (rapid growth outstrips blood supply)
Dysplasia = disordered growth with cytologic changes; NOT synonymous with neoplasia, but considered a pre-neoplastic change, particularly in epithelia. Severe dysplasia = carcinoma in situ (full-thickness involvement without basement membrane invasion).
3.2 Local Invasion
- Benign tumors grow as cohesive expansile masses, develop a fibrous capsule, and remain localized. They do NOT invade surrounding tissue.
- Malignant tumors invade adjacent tissues. They lack a well-formed capsule. Some may appear encapsulated but microscopy reveals invasion. Even a thin rim of fibrous tissue does not guarantee benignancy.
3.3 Metastasis
- Benign tumors do NOT metastasize.
- Malignant tumors have the capacity to metastasize; this is the single most important feature distinguishing benign from malignant.
- ~30% of newly diagnosed cancer patients have clinically detectable metastases; another 20% have occult metastases at the time of diagnosis.
Routes of metastasis:
- Seeding of body cavities - e.g., ovarian carcinoma spreading through the peritoneal cavity, mesothelioma seeding the pleural space
- Lymphatic spread - most common for carcinomas; follows natural lymphatic drainage. Sentinel lymph node = first node to receive lymph drainage from the primary tumor; biopsy used to stage cancer
- Hematogenous spread - more common for sarcomas; veins are penetrated more easily than arteries. Portal drainage → liver; systemic venous drainage → lungs; vertebral plexus → vertebral column
Exceptions: Some tumors preferentially metastasize to specific organs beyond what lymphatic/vascular anatomy would predict (organ tropism) - likely due to expression of receptors that match chemokines/adhesion molecules at the target organ.
4. EPIDEMIOLOGY
- Worldwide (2018): >17 million cancer cases; 9.6 million deaths (~26,300/day)
- US (2022 estimate): 1.9 million new cases; 609,000 deaths
- Death rates have decreased ~20% in men and ~10% in women since 1995
- In men: decreased lung, prostate, colon cancer deaths (80% of decrease)
- In women: decreased breast and colorectal cancer deaths (60% of decrease)
Environmental Factors
- Dominant risk factors for many common cancers
- Geographic variation in cancer rates reflects different environmental exposures
- Key factors: tobacco (most important), diet, alcohol, obesity, infectious agents, radiation, chemical carcinogens
Age
- Cancer risk increases with age, mostly due to accumulation of somatic mutations
- Some cancers occur predominantly in children (e.g., ALL, nephroblastoma/Wilms tumor, retinoblastoma, neuroblastoma)
Acquired Predisposing Conditions
- Chronic inflammation - increases cancer risk (e.g., H. pylori → gastric cancer; hepatitis B/C → hepatocellular carcinoma; Barrett esophagus → esophageal adenocarcinoma; inflammatory bowel disease → colorectal carcinoma)
- Precancerous conditions (increased risk but not inevitable):
- Chronic atrophic gastritis
- Solar keratosis of the skin
- Ulcerative colitis
- Leukoplakia of oral cavity/vulva/penis
- Villous adenomas of the colon
- Excessive hormonal stimulation - e.g., unopposed estrogen → endometrial carcinoma
5. GENETIC LESIONS IN CANCER
5.1 Driver vs. Passenger Mutations
- Driver mutations - provide growth advantage; directly contribute to cancer development
- Passenger mutations - "neutral" hitchhiker mutations that don't provide growth advantage
- Important: passenger mutations can become driver mutations if selective pressure changes (e.g., during drug treatment)
5.2 Types of Genetic Lesions
| Type | Mechanism | Effect |
|---|---|---|
| Point mutations | Single nucleotide change | Constitutive activation (e.g., RAS) or loss of function (e.g., TP53) |
| Gene rearrangements/translocations | Chromosomal rearrangements | Oncogene overexpression OR novel fusion proteins |
| Deletions | Loss of chromosomal region | Loss of tumor suppressor genes |
| Gene amplifications | Extra copies of gene | Increased oncogene expression (e.g., HER2, MYC, MYCN) |
| Aneuploidy | Abnormal chromosome number | Complex genomic instability |
5.3 MicroRNAs (miRNAs) and Cancer
- miRNAs are small non-coding RNAs that regulate gene expression post-transcriptionally
- Overexpression of miRNA → reduced expression of tumor suppressor genes
- Loss of miRNA expression → overexpression of oncogenes
- miR-21 is overexpressed in many tumors; miR-15a/miR-16-1 are deleted in many CLL cases
5.4 Epigenetic Modifications
- DNA methylation of CpG islands in promoters → gene silencing
- Histone modifications alter chromatin structure
- Tumor suppressor genes may be silenced by epigenetic changes (even without mutation)
- Implication: epigenetic silencing is potentially reversible - therapeutic target
6. CARCINOGENESIS: A MULTISTEP PROCESS
- Cancer arises from the progressive accumulation of mutations in multiple genes
- Tumors evolve through clonal expansion with each successive mutation providing growth advantage
- Two key concepts:
- Initiation - initial DNA damage creating a mutated cell
- Promotion - subsequent stimulation of initiated cells to proliferate
7. HALLMARKS OF CANCER
(Hanahan & Weinberg framework - 8 hallmarks + 2 enabling characteristics)
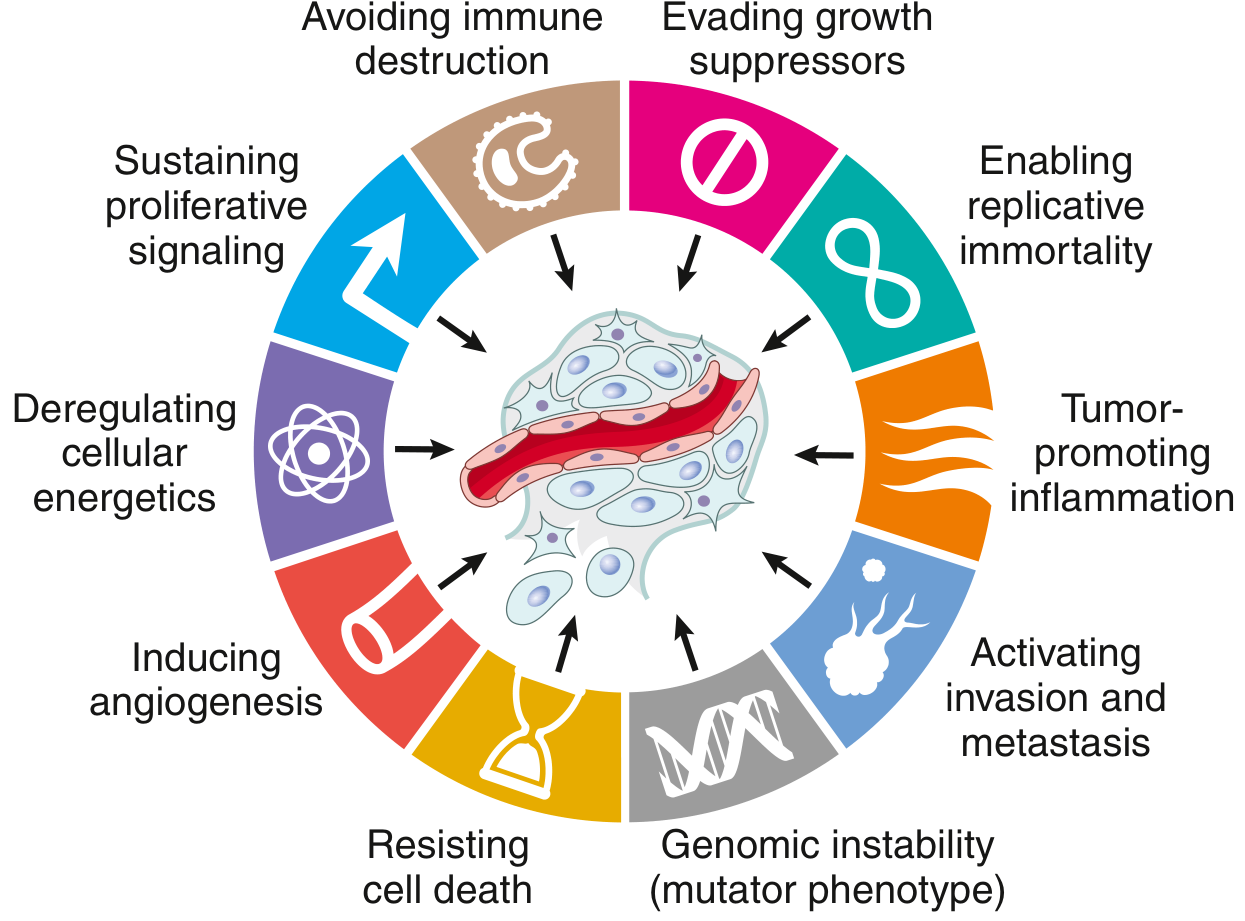
7.1 Self-Sufficiency in Growth Signals
Normal cell proliferation requires external growth factor → receptor → signal transduction → transcription factor → cell cycle entry.
Oncogenes = mutant or overexpressed versions of proto-oncogenes; produce oncoproteins that drive proliferation without external signals.
Mechanisms of oncogene activation:
a) Growth Factors - Autocrine signaling
- Tumor cells secrete their own growth factors (e.g., PDGF in brain tumors, TGF-α in some carcinomas)
- Creates autocrine loop
b) Growth Factor Receptors
- Mutations cause constitutive receptor activity (no ligand needed)
- HER2 (ERBB2) - amplified/overexpressed in 20-25% of breast cancers → constitutively active tyrosine kinase → targeted by trastuzumab (Herceptin)
- EGFR (HER1) - overexpressed in lung and other carcinomas
- FLT3 - mutated in acute myeloid leukemia
c) Signal-Transducing Proteins
- RAS - most commonly mutated oncogene in human cancers (~30% of all cancers)
- RAS is a GTP-binding protein; active when GTP-bound, inactive when GDP-bound
- Mutations (codons 12, 13, 61) impair GTPase activity → permanently GTP-bound → constitutively active
- Activates downstream: RAF/MAPK pathway and PI3K/AKT pathway
- BCR-ABL (CML) - translocation t(9;22) creates Philadelphia chromosome; BCR-ABL fusion protein has constitutively active tyrosine kinase activity → targeted by imatinib (Gleevec)
- BRAF - mutated in ~60% of melanomas; targeted by vemurafenib
d) Transcription Factors
- MYC - regulates expression of many growth-promoting genes; overexpressed or amplified in many cancers (Burkitt lymphoma: t(8;14) MYC translocation)
- MYCN - amplified in neuroblastoma
e) Cyclins and CDKs (Cell Cycle)
- Cell cycle progression controlled by cyclin-CDK complexes
- CDK inhibitors (CDKIs) normally brake cell cycle at G1/S and G2/M checkpoints
- Cyclin D - overexpressed in breast, esophageal, and other cancers → drives cells past G1/S checkpoint
- CDK4 - amplified in some sarcomas
7.2 Insensitivity to Growth Inhibitory Signals: Tumor Suppressor Genes
Tumor suppressors normally apply brakes to cell proliferation. Their loss removes these constraints. Two-hit hypothesis (Knudson): both alleles must be inactivated.
RB Gene (Retinoblastoma Gene) - "Governor of the Cell Cycle"
- Location: 13q14
- Protein: pRB - critical regulator of G1/S transition
- Mechanism: Hypophosphorylated pRB binds and sequesters E2F transcription factors; when phosphorylated by cyclin D-CDK4/6 complex → releases E2F → cell enters S phase
- In cancer: Loss of RB means E2F is always free → uncontrolled S phase entry
- Clinical associations:
- Retinoblastoma: inherited (germline 1 hit + somatic 1 hit) or sporadic (2 somatic hits)
- Also lost in osteosarcoma, breast, lung, bladder cancers
TP53 Gene - "Guardian of the Genome"
- Most commonly mutated gene in human cancers (majority of cancers)
- Location: 17p13
- Protein: p53 - transcription factor activated by cellular stress
p53 functions:
- Cell cycle arrest at G1 (via p21 induction → CDK inhibition → RB stays active)
- DNA damage repair gene induction
- Senescence induction (permanent G1 arrest with specific chromatin changes)
- Apoptosis (via upregulation of proapoptotic genes e.g., BAX)
p53 regulation:
- In normal cells: p53 levels low due to MDM2 ubiquitin-mediated degradation
- On DNA damage: kinases phosphorylate p53 → cannot be degraded by MDM2 → p53 accumulates
- After successful repair: p53 upregulates MDM2 → destroys itself → cell cycle resumes
- Li-Fraumeni syndrome: germline TP53 mutation → multiple early-onset cancers
Other Tumor Suppressors:
| Gene | Protein | Cancer |
|---|---|---|
| APC | Adenomatous polyposis coli | Colorectal cancer; familial adenomatous polyposis |
| BRCA1/BRCA2 | DNA repair | Breast and ovarian cancer |
| VHL | Von Hippel-Lindau | Renal cell carcinoma |
| PTEN | Phosphatase | Endometrial, prostate, glioblastoma |
| CDKN2A (p16) | CDK inhibitor | Melanoma, pancreatic cancer |
| NF1, NF2 | Neurofibromin | Neurofibromatosis types 1 and 2 |
| WT1 | Wilms tumor gene | Nephroblastoma |
| DPC4/SMAD4 | TGF-β signaling | Pancreatic cancer |
7.3 Altered Cellular Metabolism (Warburg Effect)
- Normal cells: aerobic respiration (oxidative phosphorylation)
- Cancer cells: predominantly use aerobic glycolysis (glycolysis even in the presence of O2) - the Warburg effect
- Glycolysis produces less ATP per glucose but:
- Provides rapid energy
- Generates biosynthetic intermediates for building new cells
- Reduces oxidative damage
- Clinical application: Basis for FDG-PET scanning (cancer cells take up more 18F-FDG due to upregulated glucose transporters)
- Oncometabolism: IDH1/IDH2 mutations (found in gliomas, AML) produce 2-hydroxyglutarate, an oncometabolite that drives epigenetic changes
- Autophagy: Cancer cells may exploit autophagy to survive nutrient deprivation; complex dual role (suppressive early, supportive later)
7.4 Evasion of Apoptosis
Apoptosis pathways:
- Intrinsic (mitochondrial) pathway: Regulated by BCL2 family (proapoptotic: BAX, BAK; antiapoptotic: BCL2, BCL-XL, MCL1)
- Extrinsic (death receptor) pathway: FAS ligand binds FAS receptor → caspase-8 activation
Cancer mechanisms to evade apoptosis:
- BCL2 overexpression - most common; t(14;18) in follicular lymphoma brings BCL2 under Ig promoter → constitutive BCL2 expression → apoptosis resistance
- Loss of TP53 - removes apoptosis trigger
- Upregulation of survival signals (PI3K/AKT pathway)
- Loss of FAS or FAS-L receptor expression
7.5 Limitless Replicative Potential (Immortality)
- Normal cells: limited cell divisions due to progressive telomere shortening → senescence or apoptosis ("Hayflick limit" ~60-70 divisions)
- Telomeres: TTAGGG repeats at chromosome ends; maintained by telomerase
- Normal somatic cells: telomerase inactive → telomeres shorten with each division
- Cancer cells: reactivate telomerase (TERT) → maintain telomere length → unlimited replication
- Telomerase expression found in ~90% of human cancers
- HPV E6 protein stimulates TERT expression
7.6 Sustained Angiogenesis
- Tumors >1-2mm need a blood supply; can't rely on diffusion
- Normal angiogenesis is tightly regulated by pro- and anti-angiogenic signals
- Cancer: tips the balance toward angiogenesis
Key angiogenic factors:
- VEGF (Vascular Endothelial Growth Factor) - main driver of tumor angiogenesis; induced by hypoxia (HIF-1α pathway), RAS activation, loss of p53
- bFGF (Basic Fibroblast Growth Factor)
- Angiopoietins - stabilize/destabilize vessels
Anti-angiogenic factors (normally suppress angiogenesis):
- Thrombospondin-1 (TSP-1) - induced by p53; lost when p53 is mutated
- Angiostatin, Endostatin
Angiogenic switch: When pro-angiogenic signals overwhelm anti-angiogenic signals, tumor switches to angiogenic phenotype. MMP-9 releases VEGF from ECM-sequestered pools.
Tumor vessels are abnormal - leaky, tortuous, poorly perfused.
Clinical application: Anti-VEGF therapy (bevacizumab) approved for multiple cancers.
7.7 Invasion and Metastasis
Steps of the Metastatic Cascade:
1. Invasion of ECM:
- Loosening of intercellular connections: Reduced E-cadherin expression (via mutation, SNAIL/TWIST transcription factors, or β-catenin activation) → cells detach
- Local ECM degradation: Tumor cells or recruited stromal cells secrete proteases:
- Matrix metalloproteinases (MMPs) - especially MMP-2 and MMP-9 (gelatinases)
- Cathepsin D, urokinase plasminogen activator
- MMP-9 cleaves type IV collagen in basement membranes; also releases ECM-sequestered VEGF
- Altered cell-ECM attachment: Integrins change expression → tumor cells can migrate
- Locomotion: Chemotactic gradients (from degraded ECM, stroma, and inflammatory cells) guide tumor cell migration via actin cytoskeleton remodeling
2. Vascular Dissemination:
- Tumor cells enter blood vessels (intravasation)
- In circulation, protected by platelets (form emboli)
- Arrest in capillary beds (size-dependent or receptor-mediated)
- Extravasation at distant site
- Most circulating tumor cells die - metastasis is an INEFFICIENT process
3. Formation of Metastases:
- Dormant micrometastases may persist for years
- Growth requires angiogenesis and immune evasion at new site
- Organ tropism: Chemokines/adhesion molecules at target organ match receptors on tumor cells
Epithelial-Mesenchymal Transition (EMT):
- Epithelial cancer cells downregulate E-cadherin and other epithelial markers
- Upregulate vimentin, N-cadherin, fibronectin (mesenchymal markers)
- Gain migratory/invasive phenotype
- Driven by SNAIL, TWIST, ZEB1/2 transcription factors
7.8 Evasion of Immune Surveillance
Anti-tumor immune responses:
- CD8+ cytotoxic T lymphocytes (CTLs) - main effectors; recognize tumor-associated antigens on MHC class I
- NK cells - kill cells lacking MHC class I
- Macrophages (M1 phenotype) - direct cytotoxicity
- Antibody-dependent cellular cytotoxicity (ADCC)
Tumor antigens:
- Products of mutated oncogenes/tumor suppressors (neoantigens) - most immunogenic
- Overexpressed normal proteins (e.g., HER2)
- Cancer-testis antigens (MAGE, GAGE) - normally expressed only in testis
- Viral antigens (e.g., HPV E6/E7, EBV antigens)
Immune evasion mechanisms:
- Downregulation of MHC class I → invisible to CTLs
- Loss of tumor antigens (antigen loss variants)
- Expression of immune checkpoint molecules:
- PD-L1 on tumor cells binds PD-1 on T cells → T cell exhaustion/anergy
- CTLA-4 on T cells → dampens T cell activation
- Secretion of immunosuppressive cytokines (TGF-β, IL-10)
- Recruitment of regulatory T cells (Tregs) and M2 macrophages into tumor microenvironment
Clinical application - Checkpoint inhibitors:
- Anti-PD-1 (pembrolizumab, nivolumab): approved for melanoma, NSCLC, and many others
- Anti-PD-L1 (atezolizumab, durvalumab)
- Anti-CTLA-4 (ipilimumab): approved for melanoma
- Revolutionary breakthrough in cancer therapy
Enabling Characteristics:
Genomic Instability:
- Accelerates accumulation of driver mutations
- Caused by: defective DNA repair, impaired mitotic checkpoints, telomere crisis
- Microsatellite instability (MSI): Defective DNA mismatch repair (MLH1, MSH2, MSH6, PMS2) → Lynch syndrome; also sporadic colorectal, endometrial cancers
- Chromosomal instability (CIN): Aberrant mitotic spindle checkpoints → aneuploidy
Tumor-Promoting Inflammation:
- Inflammatory cells provide growth factors, survival factors, angiogenic factors, enzymes that promote invasion
- Chronic inflammation creates a permissive microenvironment for tumor progression
8. CARCINOGENIC AGENTS
8.1 Chemical Carcinogens
Direct-acting agents (do NOT require metabolic activation):
- Alkylating agents (e.g., cyclophosphamide - therapeutic irony: used to treat cancer, can cause secondary AML)
- Acylating agents
Indirect-acting agents (Procarcinogens) - require metabolic activation by cytochrome P-450:
- Polycyclic aromatic hydrocarbons (PAH) - in coal tar, tobacco smoke → lung, skin, bladder cancer
- Aromatic amines / azo dyes - β-naphthylamine → bladder cancer
- Aflatoxin B1 (from Aspergillus) - hepatocellular carcinoma; common in Africa/Asia
- Nitrosamines - gastric cancer
- Benzene - AML
Steps of chemical carcinogenesis:
- Initiation - rapid, irreversible, subthreshold mutational damage to DNA
- Promotion - repeated exposure to promoter agents (not carcinogenic alone) → proliferation of initiated cells
- Progression - acquisition of additional mutations → malignant phenotype
8.2 Radiation
- Ionizing radiation: Causes DNA strand breaks, chromosomal aberrations, point mutations
- UV radiation → skin cancer (squamous cell carcinoma, basal cell carcinoma, melanoma); causes pyrimidine dimers (CC→TT signature mutations) → impairs DNA repair (nucleotide excision repair)
- X-rays, gamma rays → thyroid, breast, leukemia (AML > ALL; CLL NOT caused by radiation)
- Japanese atomic bomb survivors → leukemia, thyroid, breast cancers
- Xeroderma pigmentosum: autosomal recessive; defective NER → extreme UV sensitivity → multiple skin cancers
8.3 Oncogenic Viruses and Microbes
DNA Viruses:
| Virus | Cancer |
|---|---|
| HPV 16, 18 (high-risk) | Cervical cancer, oropharyngeal cancer, anogenital cancers |
| HPV 6, 11 (low-risk) | Condyloma (benign warts) |
| EBV | Burkitt lymphoma, nasopharyngeal carcinoma, primary CNS lymphoma (in immunocompromised), Hodgkin lymphoma |
| HBV | Hepatocellular carcinoma (via chronic inflammation + HBx protein) |
| KSHV (HHV-8) | Kaposi sarcoma |
| Merkel cell polyomavirus | Merkel cell carcinoma |
HPV Mechanisms:
- E6 protein: binds and degrades p53; stimulates TERT (telomerase) → immortalization
- E7 protein: binds RB → releases E2F → cell cycle progression; inactivates p21
- High-risk HPV types have higher affinity for p53 and RB than low-risk types
- Integration into host genome → overexpression of E6/E7 + genomic instability
RNA Viruses:
- HTLV-1 → Adult T-cell leukemia/lymphoma (ATL); Tax protein activates NF-κB → proliferation
H. pylori:
- Chronic gastritis → gastric adenocarcinoma and MALT lymphoma
- Chronic inflammation drives carcinogenesis
9. CLINICAL ASPECTS OF NEOPLASIA
9.1 Effects of Tumor on the Host
-
Local effects (compression, invasion, obstruction):
- Small pituitary adenoma → hypopituitarism
- Small leiomyoma near renal artery → renal ischemia, hypertension
- Small bile duct carcinoma → fatal biliary obstruction
- Tumors in gut lumen → intussusception, obstruction
-
Hormonal effects:
- Pancreatic islet cell adenoma/carcinoma → hyperinsulinism
- Adrenocortical adenoma/carcinoma → hyperaldosteronism (hypertension, hypokalemia)
- Better differentiated tumors more likely to secrete hormones
-
Cachexia:
- Progressive loss of body fat + lean body mass; profound weakness, anorexia, anemia
- NOT caused by nutritional demands of the tumor
- Mediated by cytokines: TNF (suppresses appetite, inhibits lipoprotein lipase), IL-1, IL-6, IFN-γ
- Basal metabolic rate INCREASED (unlike starvation, where BMR decreases)
- No effective treatment except removing the tumor
9.2 Paraneoplastic Syndromes
- Symptoms NOT explained by local/distant spread or by normal tissue hormone production
- Occur in 10-15% of cancer patients
- Important because:
- May be first manifestation of occult cancer
- May be clinically significant or lethal
- May mimic metastasis (confound treatment)
| Syndrome | Associated Cancer | Mechanism |
|---|---|---|
| Hypercalcemia | Squamous cell Ca lung, breast Ca, renal Ca, adult T-cell lymphoma | PTHrP (PTH-related peptide); osteolytic metastases; ectopic PTH |
| Cushing syndrome | Small cell lung Ca, neural tumors | Ectopic ACTH production |
| SIADH | Small cell lung Ca, intracranial neoplasms | Ectopic ADH production |
| Non-bacterial thrombotic endocarditis | Advanced mucin-secreting carcinomas | Hypercoagulable state |
| Trousseau phenomenon | Pancreatic Ca, bronchogenic Ca, others | Migratory thrombophlebitis; procoagulants from tumor |
| Polycythemia | Renal Ca, hepatocellular Ca, cerebellar hemangioblastoma | Ectopic EPO |
| Hypoglycemia | Fibrosarcoma, hepatocellular Ca | IGF-2; ectopic insulin |
| Acanthosis nigricans | Gastric, lung, uterine carcinomas | TGF-α, other factors |
| Hypertrophic osteoarthropathy / Clubbing | Bronchogenic carcinoma | Unknown |
| Lambert-Eaton syndrome | Small cell lung Ca | Anti-VGCC antibodies (autoimmune) |
| Cerebellar degeneration | Small cell lung Ca, breast Ca | Anti-neuronal antibodies |
| Peripheral neuropathy | Various | Anti-neuronal antibodies |
9.3 Grading and Staging
Grading = microscopic assessment of degree of differentiation and mitotic activity:
- Grade 1 = well differentiated (low grade)
- Grade 2 = moderately differentiated
- Grade 3 = poorly differentiated
- Grade 4 = undifferentiated/anaplastic (high grade)
- Different systems for different tumors (Gleason for prostate, Fuhrman for RCC, etc.)
- Grading has LESS clinical importance than staging
Staging = extent of spread of cancer; more important for prognosis and treatment:
- TNM system (most widely used):
- T = size/extent of primary tumor (T1-T4)
- N = regional lymph node involvement (N0-N3)
- M = distant metastasis (M0 = none; M1 = present)
- Also expressed as clinical stages I-IV (Stage I = localized; Stage IV = distant metastasis)
9.4 Laboratory Diagnosis
Histologic / Cytologic methods:
- Biopsy (gold standard): incisional or excisional
- Fine needle aspiration (FNA): cytology of cells
- Frozen sections: rapid intraoperative diagnosis
- Core needle biopsy: good for solid tumors
- Exfoliative cytology: Pap smear (cervix), sputum, urine, pleural/peritoneal fluid
Immunohistochemistry (IHC):
- Identifies cell lineage using antibodies to tissue-specific antigens
- Examples:
- PSA → prostate cancer
- ER/PR, HER2 → breast cancer
- CD markers (CD20 → B-cell lymphoma; CD3 → T-cell lymphoma)
- Cytokeratin → carcinoma; vimentin → sarcoma; S100 → melanoma, neural tumors
- TTF-1 → lung adenocarcinoma; CK7/CK20 patterns distinguish primary sites
- Identifies hormone receptors for targeted therapy
- Identifies metastasis of unknown primary
Flow Cytometry:
- Immunophenotyping of leukemias and lymphomas
- Cell cycle analysis (DNA ploidy; S-phase fraction)
Molecular Diagnosis:
- PCR, FISH, next-generation sequencing (NGS)
- Detect gene rearrangements (BCR-ABL, IGH rearrangements)
- Identify driver mutations for targeted therapy (EGFR, ALK, RAS, BRAF, HER2)
- Microsatellite instability testing (for Lynch syndrome, anti-PD-1 therapy eligibility)
- FISH for amplifications (HER2, MYC, MYCN)
- Methylation-specific PCR for epigenetic silencing
Tumor Markers:
- Serum proteins produced by tumor cells; used for monitoring, not diagnosis
- NOT adequate for cancer screening (low sensitivity/specificity)
| Marker | Associated Cancer |
|---|---|
| PSA (Prostate Specific Antigen) | Prostate cancer |
| CEA (Carcinoembryonic Antigen) | Colorectal, gastric, pancreatic, lung, breast carcinomas |
| AFP (Alpha-fetoprotein) | Hepatocellular carcinoma, non-seminomatous germ cell tumors |
| hCG (β-human chorionic gonadotropin) | Gestational trophoblastic disease, choriocarcinoma, non-seminomatous germ cell tumors |
| CA-125 | Ovarian carcinoma |
| CA-19-9 | Pancreatic carcinoma |
| CA-15-3 | Breast carcinoma |
| Calcitonin | Medullary thyroid carcinoma |
| Inhibin | Granulosa cell tumor (ovary) |
| Chromogranin A | Neuroendocrine tumors |
SUMMARY TABLE: Benign vs. Malignant
| Feature | Benign | Malignant |
|---|---|---|
| Differentiation | Well differentiated | Variable; may be anaplastic |
| Rate of growth | Usually slow | Often rapid |
| Capsule | Usually present | Absent |
| Borders | Well-defined | Poorly defined, irregular |
| Mitoses | Rare; normal | Frequent; may be atypical |
| Local invasion | No | Yes |
| Metastasis | Never | Frequent |
| Recurrence after excision | Rare | Possible |
| Effect on host | Often minor | Often severe |
Source: Robbins & Kumar Basic Pathology (Robbins Pathology), 9780323790185, Chapter 6 - Neoplasia
This is a shared conversation. Sign in to Orris to start your own chat.