Paraganglioma
Reading File
Finding Sources
Searching PubMed
paraganglioma
Reading File
Reading File
Reading File
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Loading Image
I now have comprehensive information. Let me compile the full response.
Paraganglioma
Definition and Origin
Paragangliomas (PGLs) are neuroendocrine tumors arising from neuroectodermal cells (paraganglia) associated with the sympathetic and parasympathetic nervous systems. They occur at many sites throughout the body, from the skull base to the pelvis. The adrenal medullary pheochromocytoma is the most common paraganglioma, accounting for 80-85% of all cases - the term "paraganglioma" is increasingly reserved for extra-adrenal tumors. Approximately 70% of extra-adrenal paragangliomas occur in the head and neck.
- Robbins, Cotran & Kumar Pathologic Basis of Disease, p. 694
Classification by Location
1. Sympathetic Paragangliomas
- Arise from paravertebral paraganglia (e.g., organ of Zuckerkandl - aortic bifurcation)
- Stain positively for chromaffin (cells producing catecholamines)
- More likely to be functional (secreting catecholamines)
2. Parasympathetic Paragangliomas (Head and Neck)
These arise near the great vessels and cranial nerves, and only rarely produce catecholamines:
- Carotid body tumor - most common; arises at the bifurcation of the common carotid artery
- Glomus jugulare (jugulotympanic) - arises from the jugular bulb; can fill the middle ear
- Glomus tympanicum - arises from the cochlear promontory
- Vagal paraganglioma - from the ganglion nodosum of the vagus nerve
- Laryngeal paraganglioma - 3rd most common neuroendocrine tumor of the larynx; 3x more common in women
Distribution in head/neck: ~50% temporal bone, ~35% carotid body, ~12% high cervical vagus.
- Scott-Brown's Otorhinolaryngology Head & Neck Surgery
Histopathology
The hallmark microscopic feature is the Zellballen pattern - nests of round-to-oval chief cells surrounded by delicate fibrovascular septa. Two cell types are present:
- Chief cells (type I): neuroectodermal origin; abundant clear or granular eosinophilic cytoplasm; uniform round-to-ovoid nuclei. Stain positively for: chromogranin, synaptophysin, INSM1 (insulinoma-associated protein 1), and CD56
- Sustentacular cells (type II): spindle-shaped supporting cells at the periphery of nests; positive for S-100 protein
Little cellular pleomorphism and scant mitoses are typical. Electron microscopy may reveal neuroendocrine granules, especially in sympathetic tumors.
- Robbins, Cotran & Kumar Pathologic Basis of Disease, p. 694
Histology (Carotid body tumor):
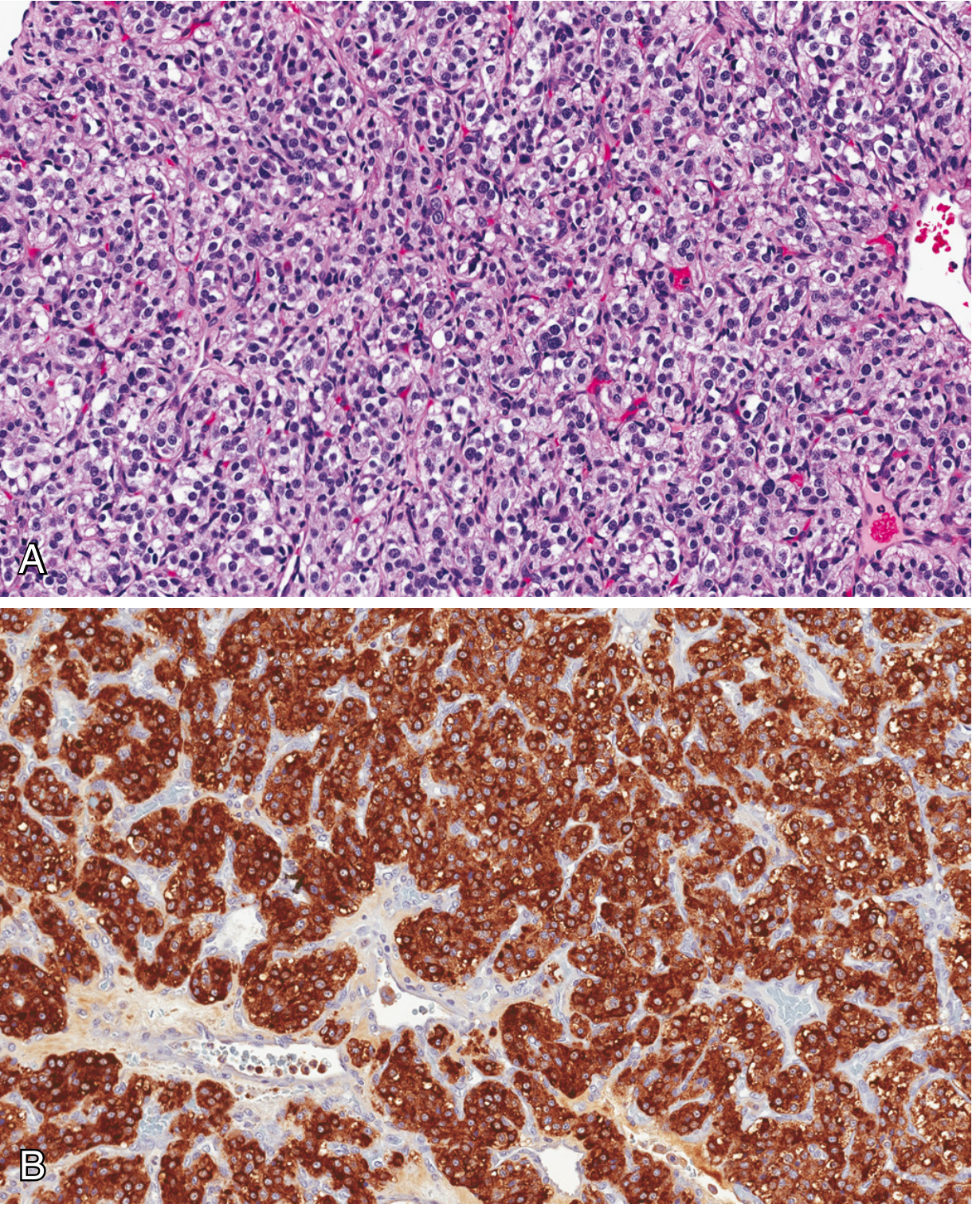
Genetics
Paragangliomas have one of the highest rates of heritable mutations among all tumors. Key genes:
| Gene | Locus | Association |
|---|---|---|
| SDHB | 1p36 | Highest malignancy risk (30-50%); abdominal/thoracic PGL |
| SDHD | 11q23 | Head/neck PGL; maternal imprinting - only penetrant if inherited paternally |
| SDHC | 1q21 | Head/neck PGL |
| SDHAF2 | 11q13 | Head/neck PGL; maternal imprinting; loss of FAD cofactor attachment |
| SDHA | 5p15 | Rare; gastric GIST-PGL syndrome |
| NF1 | 17q11 | Neurofibromatosis type 1 (~3% develop pheo/PGL) |
| RET | 10q11 | MEN2A/2B - usually adrenal pheo |
| VHL | 3p25 | Von Hippel-Lindau - abdominal PGL |
Loss-of-function mutations in SDH genes impair mitochondrial complex II, likely promoting tumorigenesis by altering cellular metabolism (pseudohypoxia signaling via HIF). ~1/3 of head/neck PGLs have germline mutations. Familial PGL is autosomal dominant.
- Goldman-Cecil Medicine, Sabiston Textbook of Surgery, Robbins
Clinical Features
General characteristics:
- Rare, slow-growing, usually painless masses
- Peak incidence: 5th-6th decades
- Incidence is higher at high altitudes (carotid body tumors - chronic hypoxia)
- ~10% of patients have multifocal tumors
- ~5% of head/neck PGLs secrete catecholamines (vs. ~50% of sympathetic PGLs)
Symptoms by location:
- Carotid body tumor: painless neck mass at the carotid bifurcation; Fontaine sign (limited vertical mobility, some lateral mobility); may cause cranial nerve palsies
- Glomus tympanicum: pulsatile tinnitus; red pulsatile mass visible behind intact tympanic membrane ("rising sun" sign)
- Glomus jugulare: pulsatile tinnitus + conductive hearing loss; cranial nerve IX, X, XI palsies (jugular foramen syndrome)
- Vagal PGL: neck mass with vagal palsy (hoarseness, dysphagia)
Catecholamine excess symptoms (when secretory):
Episodic paroxysms of hypertension, forceful heartbeat, pallor, tremor, headache, and diaphoresis. The classic triad is headache, palpitations, and sweating. Spells last ~15-20 minutes and can be triggered by positional change, medications (metoclopramide, beta-blockers, anesthetic agents), or increased intra-abdominal pressure.
- Goldman-Cecil Medicine, Cummings Otolaryngology
Malignancy
- Histologic features (mitoses, pleomorphism, vascular invasion) do not reliably predict malignancy
- Malignancy is defined by the presence of metastases in sites where paraganglionic tissue is not normally found (e.g., lymph nodes, liver, lung, bone)
- SDHB mutations carry the highest metastatic risk: 30-50%
- Up to 50% of metastatic PGLs are ultimately fatal, largely due to infiltrative growth
- Carotid body tumors may metastasize to regional lymph nodes despite benign appearance
- Robbins, p. 694-695
Imaging and Diagnosis
- CT (high-resolution): best for bony involvement (skull base erosion in glomus jugulare)
- MRI: best for soft tissue extent, intracranial invasion, and vascular encasement; classic "salt and pepper" appearance on T2 due to signal voids from flow
- Octreotide scan / 68Ga-DOTATATE PET: somatostatin receptor scintigraphy - highly sensitive for functional imaging
- Angiography: enlarged feeding arteries with intense early tumor blush; used pre-embolization; confirms centripetal arterioles with arteriovenous shunts
- Biopsy warning: highly vascular - pre-biopsy imaging to confirm diagnosis first; risk of severe hemorrhage. Radiographic evidence is often sufficient for diagnosis.
- Biochemistry: 24-hr urinary/plasma metanephrines and catecholamines in secretory tumors
Treatment
Treatment options include observation, surgery, radiation, and targeted therapy. Decision-making incorporates patient age, comorbidities, tumor size/location, and patient preference.
- Cummings Otolaryngology, p. (block34)
Surgery:
- Treatment of choice for most resectable tumors
- Glomus tympanicum: amenable to excision with minimal morbidity; ~80-95% success
- Glomus jugulare: significant risk to cranial nerves VII, IX, X, XI
- Carotid body tumor: risk of stroke and cranial nerve injury
- Preoperative embolization (24-48 hrs before surgery): reduces intraoperative blood loss; particularly for temporal bone and large carotid body PGLs; recent meta-analysis (PMID: 38942141) assessed its role in jugular PGL surgery
- Laryngeal PGL: lateral thyrotomy/pharyngotomy preferred; transoral laser discouraged due to hemostasis difficulties
Radiation:
- External beam radiation and stereotactic radiosurgery (Gamma Knife) both used
- As primary modality: halts tumor growth in >90% of cases but may not relieve symptoms
- Also used as adjuvant for residual or recurrent disease
- Responds but incompletely to RT
Medical therapy (secretory tumors):
- Pre-operative alpha-blockade (phenoxybenzamine or doxazosin) MANDATORY before surgery
- Beta-blockade added ONLY after adequate alpha-blockade
- For metastatic/unresectable: sunitinib, MIBG therapy (MIBG-avid tumors), CVD chemotherapy (cyclophosphamide, vincristine, dacarbazine), peptide receptor radionuclide therapy (PRRT) with 177Lu-DOTATATE for SSTR-positive tumors
Syndromes with Paraganglioma
| Syndrome | Gene | Key Features |
|---|---|---|
| Familial PGL type 1 | SDHD | Head/neck PGL; maternal imprinting |
| Familial PGL type 2 | SDHAF2 | Head/neck PGL; maternal imprinting |
| Familial PGL type 3 | SDHC | Head/neck PGL |
| Familial PGL type 4 | SDHB | Abdominal/thoracic PGL; high malignancy |
| MEN2A/2B | RET | Bilateral adrenal pheo |
| Von Hippel-Lindau | VHL | Abdominal PGL + retinal hemangioma + renal cell Ca + CNS hemangioblastoma |
| Neurofibromatosis 1 | NF1 | Usually adrenal pheo (~3%) |
| Carney triad | Unknown | GIST + pulmonary chondroma + PGL |
Key Concepts Summary
- Paragangliomas = neuroendocrine tumors of paraganglia; pheochromocytoma is the adrenal subtype
- 70% of extra-adrenal PGLs are in the head/neck (parasympathetic, rarely secretory)
- Zellballen architecture; chief cells positive for chromogranin/synaptophysin; sustentacular cells positive for S-100
- Highly heritable: SDH gene mutations in majority of hereditary cases; SDHB = highest malignancy risk
- Malignancy defined only by metastasis - not by histologic features
- Preoperative embolization for vascular control; mandatory alpha-blockade before surgery if secretory
- SDHB pathogenic variant outcomes - systematic review (2024), PMID 38605204
- Preoperative embolization in jugular PGL surgery - meta-analysis (2024), PMID 38942141
This is a shared conversation. Sign in to Orris to start your own chat.