Teach. Me everything about ADH in simplified manner
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Reading File
Finding Sources
Reading File
Loading Image
Finding Sources
Loading Image
I have all the information needed. Here is a complete, simplified guide to ADH:
ADH (Antidiuretic Hormone) - Everything You Need to Know
1. What IS ADH?
ADH, also called vasopressin or arginine vasopressin (AVP), is a small peptide hormone (a nonapeptide - just 9 amino acids) made in the hypothalamus and stored in + released from the posterior pituitary gland.
Think of it as your body's "water saver" - its main job is to stop you from losing too much water in your urine.
- Katzung's Basic and Clinical Pharmacology, 16th Ed.
2. Where is it Made and Stored?
| Location | Role |
|---|---|
| Supraoptic nucleus (hypothalamus) | Main site of ADH synthesis |
| Paraventricular nucleus (hypothalamus) | Also produces ADH |
| Posterior pituitary | Stores and releases it into blood |
The neurons in the hypothalamus literally send their axons down into the posterior pituitary - so the hormone is made in the brain but released from the pituitary.
3. What Triggers its Release?
Two major stimuli:
A. High Blood Osmolality (most important)
- Osmolality = how concentrated your blood is (basically how much sodium/solutes are dissolved)
- Normal blood osmolality = ~285-290 mOsm/L
- When you're dehydrated or eat salty food, osmolality rises
- Osmoreceptors in the hypothalamus detect this and signal ADH release
B. Low Blood Volume / Low Blood Pressure
- When you lose blood or fluids (e.g., hemorrhage, vomiting), baroreceptors in the aorta/carotid detect the drop
- They signal the hypothalamus to release ADH
- This is a stronger emergency signal - even small drops in BP cause big ADH surges
Other stimuli that INCREASE ADH:
- Nausea/vomiting
- Pain, stress, surgery
- Hypoxia (low oxygen)
- Certain drugs (morphine, nicotine, carbamazepine, SSRIs)
Things that DECREASE ADH:
- Alcohol (this is why alcohol makes you urinate a lot)
- Atrial natriuretic peptide (ANP)
- Low osmolality (drinking lots of water)
4. How Does ADH Work? (Mechanism of Action)
ADH acts on two types of receptors:
V2 Receptors (kidney - main effect)
Located on the principal cells of the collecting duct and distal tubule.
Here is the exact mechanism, step by step:
- ADH binds to the V2 receptor on the blood-side of the collecting duct cell
- V2 is a G-protein coupled receptor - activates Gs protein → adenylyl cyclase → cAMP
- cAMP triggers protein kinase A
- This causes vesicles carrying pre-formed water channels (Aquaporin-2, AQP2) to fuse with the apical (urine-side) membrane
- Water rushes from the dilute urine in the tubule, through AQP2, across the cell, and out through AQP3/AQP4 on the blood side
- Result: concentrated urine, water retained in blood
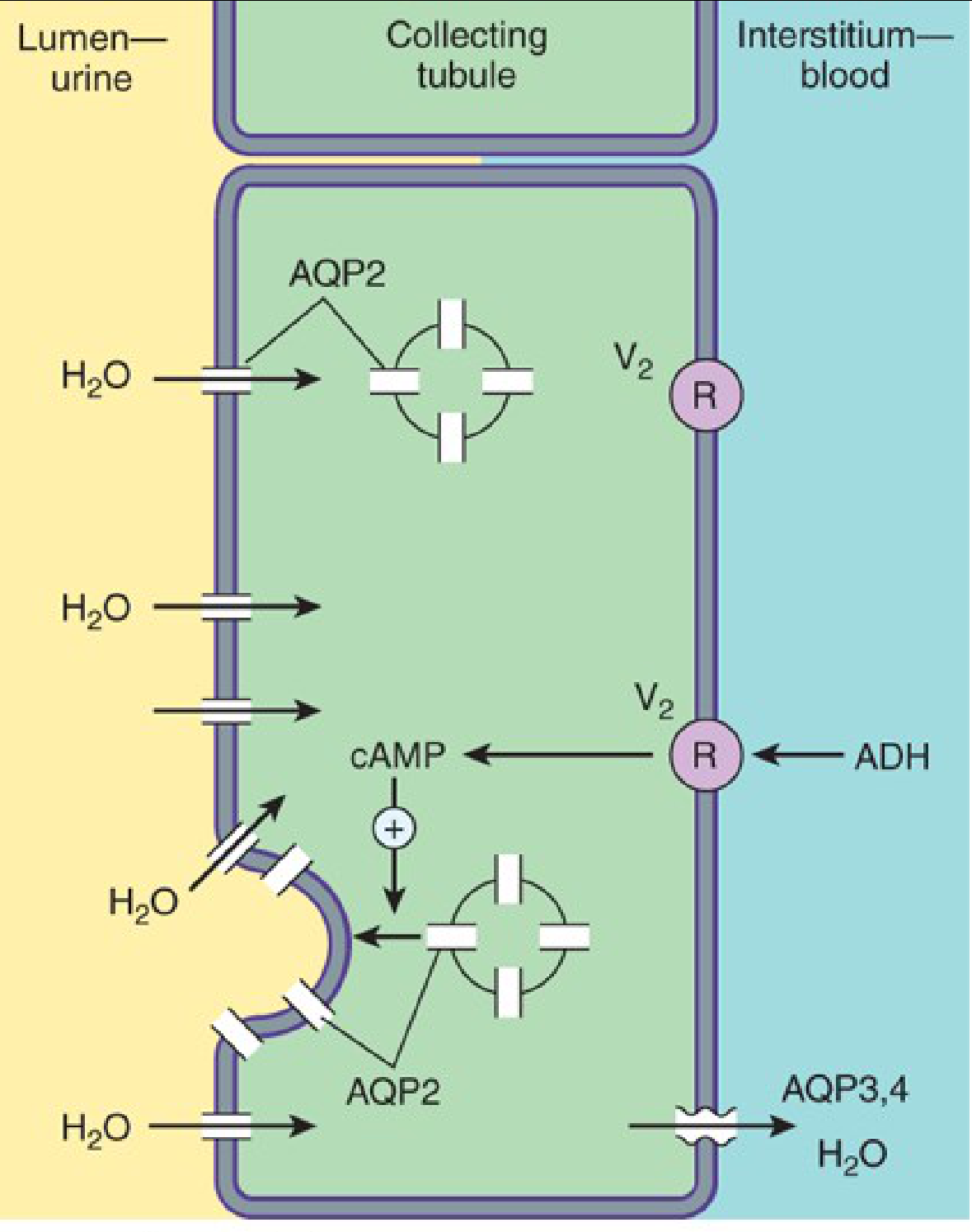
The diagram above shows exactly this process - ADH binds V2 receptor on the blood side, triggers cAMP, and AQP2 channels are inserted into the urine-side membrane, allowing water to flow out.
- Katzung's Basic and Clinical Pharmacology, 16th Ed.
V1 Receptors (blood vessels - secondary effect)
- Found on vascular smooth muscle
- Activation causes vasoconstriction (raises blood pressure)
- Coupled to Gq → phospholipase C (different pathway)
- This is why ADH is also called "vasopressin"
Extra V2-like Receptors (outside kidney)
- Stimulate release of von Willebrand factor and Factor VIII from endothelial cells
- This is why desmopressin (synthetic ADH) is used to treat bleeding disorders like hemophilia A and von Willebrand disease
5. What Happens in the Kidney? (Simplified)
Think of the nephron as a tube that processes urine in stages:
| Segment | Water permeability | What happens |
|---|---|---|
| Proximal tubule | Always permeable | Isosmotic water reabsorption (300 mOsm/L throughout) |
| Thick ascending limb of loop of Henle | Never permeable to water | NaCl reabsorbed, tubular fluid dilutes to ~100 mOsm/L |
| Early distal tubule | Not permeable | Further dilution to ~80 mOsm/L |
| Late distal + collecting duct | ADH-dependent | ADH present = water reabsorbed, urine concentrates to 1200 mOsm/L; ADH absent = dilute urine exits |
Bottom line: The collecting duct is the final "faucet" - ADH turns it on (water reabsorbed), absence of ADH turns it off (water lost in urine).
- Costanzo Physiology, 7th Ed.
6. ADH and Urea
ADH also inserts urea transporter UT-A1 into collecting duct cells in the medulla. Urea accumulates in the renal medulla, contributing to the high osmotic gradient there - this is what allows water to be pulled out by osmosis. So ADH also maintains the concentration machinery of the kidney, not just the water channels.
7. Clinical Disorders of ADH
A. Diabetes Insipidus (DI) - Too Little ADH Effect
| Type | Problem | Cause | Result |
|---|---|---|---|
| Central DI | Not enough ADH made/released | Brain tumors, head trauma, neurosurgery, congenital | Large volumes of dilute urine (polyuria + polydipsia) |
| Nephrogenic DI | Kidneys don't respond to ADH | Lithium toxicity, hypercalcemia, chronic kidney disease, genetic mutations | Same symptoms, but ADH levels are normal or high |
Both types present with:
- Polyuria (large urine output, up to 10-20 L/day)
- Polydipsia (extreme thirst)
- Hypernatremia (high blood sodium, concentrated blood)
- Dilute urine (low urine osmolality)
Treatment:
- Central DI: DDAVP (desmopressin) - synthetic ADH analog, intranasal/oral/SC
- Nephrogenic DI: low-salt diet, thiazide diuretics (paradoxically reduce urine output), demeclocycline is not used here
B. SIADH - Too Much ADH (Syndrome of Inappropriate ADH Secretion)
ADH is secreted autonomously even when blood osmolality is low - the feedback is broken.
Causes include:
- CNS disorders (meningitis, stroke, brain tumors)
- Pulmonary disease (TB, pneumonia, small cell lung cancer)
- Drugs: carbamazepine, SSRIs, NSAIDs, antipsychotics, morphine
- Post-surgical stress
What happens:
- Water is retained (dilutes the blood)
- Hyponatremia (low blood sodium) - this is the hallmark
- Concentrated urine (paradoxically, even though patient is "waterlogged")
Treatment:
- Fluid restriction (first line)
- Demeclocycline (blocks ADH action on kidneys)
- Vaptans (conivaptan, tolvaptan) - V2 receptor antagonists that block ADH's kidney effect and cause free-water excretion ("aquaresis")
8. Drugs That Target ADH
ADH Analogs (Agonists)
| Drug | Key Features | Uses |
|---|---|---|
| Vasopressin | Short half-life (~15 min), IV/IM only | Vasodilatory shock, GI variceal bleeding |
| Desmopressin (DDAVP) | Long-acting (1.5-2.5h half-life), intranasal/oral/SC, V2-selective (no pressor effect) | Central DI, nocturnal enuresis, hemophilia A, von Willebrand disease |
ADH Antagonists (Vaptans)
| Drug | Receptor selectivity | Route | Uses |
|---|---|---|---|
| Conivaptan | V1a + V2 | IV | Euvolemic or hypervolemic hyponatremia |
| Tolvaptan | V2 (30x more than V1) | Oral | Same; also polycystic kidney disease (but 30-day limit due to hepatotoxicity risk) |
Vaptans cause aquaresis - excretion of free water without sodium loss. They raise serum sodium in SIADH/heart failure/cirrhosis.
- Katzung's Basic and Clinical Pharmacology, 16th Ed.
9. Quick Reference: ADH Summary Table
| Feature | Detail |
|---|---|
| Chemical nature | Nonapeptide (9 amino acids) |
| Produced by | Supraoptic + paraventricular nuclei (hypothalamus) |
| Released from | Posterior pituitary |
| Half-life | ~15-20 minutes |
| Main trigger | High plasma osmolality / low blood volume |
| Main target | Collecting duct + distal tubule (principal cells) |
| Main receptor | V2 (kidney) - via cAMP/PKA → AQP2 insertion |
| Secondary receptor | V1 (vessels) - vasoconstriction |
| Net effect | Concentrated urine, water retention |
| Deficiency (↓ADH effect) | Diabetes Insipidus |
| Excess (↑ADH effect) | SIADH |
| Synthetic analog | Desmopressin (DDAVP) |
| Antagonists | Vaptans (conivaptan, tolvaptan) |
10. Memory Aid
"ADH = Anti-Dry Hormone" - it fights dryness/dehydration by retaining water.
The receptor shortcut:
- V1 = Vessels (vasoconstriction, Gq pathway)
- V2 = Volume/water (kidney water reabsorption, cAMP pathway)
DI vs SIADH:
- DI: Dry blood, dilute urine (too little ADH effect)
- SIADH: Dilute blood, concentrated urine (too much ADH effect)
Sources: Katzung's Basic and Clinical Pharmacology, 16th Ed. | Costanzo Physiology, 7th Ed. | Tietz Textbook of Laboratory Medicine, 7th Ed.
Explain diabetes insipidus and siadh in detail
Finding Sources
Finding Sources
Reading File
Reading File
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
I now have comprehensive material from multiple authoritative textbooks. Here is a detailed, structured explanation:
Diabetes Insipidus (DI) and SIADH - Detailed Guide
PART 1: DIABETES INSIPIDUS (DI)
The Core Concept
DI is the opposite of SIADH. The problem here is no ADH effect on the kidneys - either because ADH isn't being made, or the kidneys refuse to respond to it. The result: the kidneys cannot hold onto water, so huge volumes of dilute urine pour out.
Simple analogy: Think of the collecting duct as a tap. ADH normally closes the tap (retains water). In DI, the tap is always wide open - water pours out endlessly.
Types of DI
There are 3 main types, and understanding the difference is key:
| Type | Problem | ADH level in blood |
|---|---|---|
| Central (Neurogenic) DI | Brain doesn't make/release enough ADH | LOW |
| Nephrogenic DI | Kidneys don't respond to ADH | HIGH (body tries to compensate) |
| Primary Polydipsia (Psychogenic) | Patient drinks excessively - not a true DI | Low-normal |
1. Central (Neurogenic) DI
What happens: Destruction or damage to the hypothalamus or posterior pituitary stops ADH from being produced or released.
Important anatomical point: Lesions of the posterior pituitary alone do NOT always cause DI. The lesion has to be high enough in the pituitary stalk to cause retrograde degeneration of hypothalamic neurons in the supraoptic and paraventricular nuclei - because those are the actual ADH-producing cells. Lower stalk/pituitary lesions may spare enough ADH production.
Causes:
- Brain tumors (craniopharyngioma, pituitary adenoma, metastases)
- Head trauma
- Neurosurgery (especially pituitary surgery)
- Subarachnoid hemorrhage / intracranial hemorrhage
- Meningitis / encephalitis
- Sarcoidosis, histiocytosis X (infiltrative)
- Congenital (rare)
Special scenario - Triphasic response after pituitary surgery:
- Phase 1 (DI): Immediately post-op - damaged neurons can't release ADH → polyuria
- Phase 2 (SIADH): Days later - dying neurons dump stored ADH → water retention, hyponatremia
- Phase 3 (DI again): If enough neurons die, ADH is permanently deficient → polyuria returns
This is a classic exam and clinical scenario.
2. Nephrogenic DI
What happens: ADH is present (and even elevated), but the kidney's V2 receptors or AQP2 channels are defective or blocked.
Causes:
Genetic (congenital):
- AVPR2 gene mutations (>90% of congenital cases) - X-linked recessive, affects males predominantly, prevalence ~4 per million males
- AQP2 gene mutations (<10% of congenital cases) - can be autosomal dominant or recessive
Acquired (more common):
- Lithium - most common drug cause; blocks AQP2 insertion into cell membrane
- Demeclocycline - intentionally used to cause nephrogenic DI to treat SIADH
- Hypercalcemia (calcium interferes with cAMP signaling)
- Hypokalemia
- Chronic kidney disease
- Ureteric obstruction (downregulates AQP2)
Clinical Features of DI (Both Types)
| Symptom | Explanation |
|---|---|
| Massive polyuria | Up to 10-20 liters/day; hallmark symptom |
| Polydipsia (extreme thirst) | Compensatory - body tries to replace lost water |
| Nocturia | Night-time urination disrupts sleep |
| Hypernatremia | Blood becomes concentrated (high sodium) |
| Dilute urine | Urine osmolality very low (<300 mOsm/kg) |
Danger: If the thirst mechanism is also damaged (e.g., hypothalamic damage), the patient cannot compensate by drinking. This leads to progressive hypernatremia and brain damage from repeated hyperosmolar episodes. If DI starts in childhood, the constant high urine flow can cause massive dilation of the renal pelvis, ureters, and bladder.
Diagnosis of DI
Step 1: Rule out other causes of polyuria
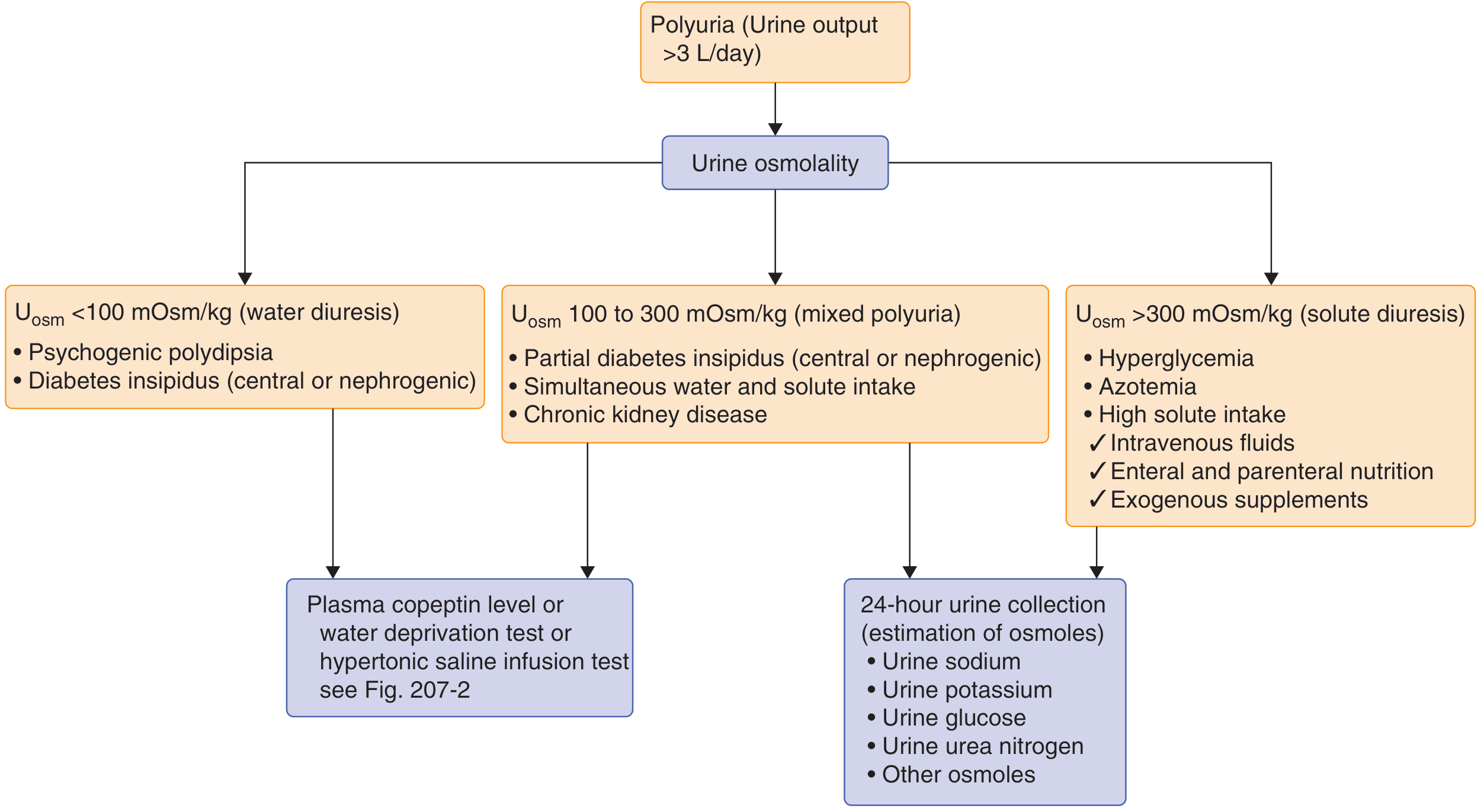
The flowchart above is your clinical guide:
- Urine osmolality <100 mOsm/kg → water diuresis (DI or psychogenic polydipsia)
- Urine osmolality 100-300 mOsm/kg → partial DI or mixed
- Urine osmolality >300 mOsm/kg → solute diuresis (diabetes mellitus, high protein diet, etc.)
Step 2: Distinguish Central DI vs Nephrogenic DI vs Primary Polydipsia
Water Deprivation Test (older gold standard):
- Deny fluids → measure urine osmolality repeatedly
- Then give desmopressin → measure again
- Central DI: Urine osmolality rises significantly after desmopressin (kidneys work fine, just needed the hormone)
- Nephrogenic DI: Urine osmolality barely rises after desmopressin (kidneys are resistant)
- Primary polydipsia: Urine concentrates normally during water deprivation
Copeptin-based test (modern, more accurate):
- Copeptin is the prohormone of ADH, has a longer half-life, easier to measure
- Baseline copeptin ≥1.4 pmol/L without water deprivation = nephrogenic DI (kidneys resistant, body compensates with high ADH)
- Copeptin >4.9 pmol/L after hypertonic saline infusion (when Na ≥150 mmol/L) = diagnoses central DI with 97% accuracy
- Harrison's Principles of Internal Medicine, 22nd Ed.
Key lab values:
| Test | Central DI | Nephrogenic DI | Primary Polydipsia |
|---|---|---|---|
| Serum Na | High or normal | High or normal | Low-normal |
| Serum osmolality | High | High | Low-normal |
| Urine osmolality | Very low | Very low | Low |
| ADH level | LOW | HIGH | Low-normal |
| Response to DDAVP | ✅ Urine concentrates | ❌ No response | Variable |
Treatment of DI
Central DI:
- Desmopressin (DDAVP) - synthetic ADH analog, first-line treatment
- Intranasal: 10-40 mcg in 2-3 divided doses
- Oral: 0.1-0.2 mg 2-3 times daily
- Subcutaneous: 1-4 mcg every 12-24 hours
- Also replace fluid losses, treat underlying cause
Nephrogenic DI:
-
Remove the cause (stop lithium if possible, correct hypercalcemia/hypokalemia)
-
Low-sodium, low-protein diet (reduces solute load, decreases obligatory urine volume)
-
Thiazide diuretics (paradoxical effect - create mild volume depletion → proximal tubule reabsorbs more water → less reaches the dysfunctional collecting duct)
-
Amiloride (especially for lithium-induced DI - blocks lithium entry into cells)
-
NSAIDs (e.g., indomethacin - block prostaglandins that normally antagonize ADH)
-
Desmopressin alone is NOT effective in nephrogenic DI
-
Brenner and Rector's The Kidney, 2-Volume Set
PART 2: SIADH (Syndrome of Inappropriate ADH Secretion)
The Core Concept
In SIADH, ADH is being secreted when it shouldn't be - the normal feedback is broken. The body has enough (or too much) water, but ADH keeps telling the kidneys to hold onto even more. The result: too much water, diluted blood, low sodium.
Analogy: SIADH is like someone who keeps closing the drain even when the bathtub is already full. Water overflows (dilutes everything).
Diagnostic Criteria for SIADH
All of these must be present:
| Criterion | Value |
|---|---|
| Hyponatremia | Serum Na <135 mEq/L |
| Serum hypo-osmolality | Plasma osmolality <275 mOsm/kg |
| Urine osmolality inappropriately elevated | >200 mOsm/kg (concentrated urine despite dilute blood) |
| Urine sodium elevated | >20 mEq/L (kidneys keep excreting Na) |
| Clinical euvolemia | No edema, no dehydration |
| Normal adrenal, thyroid, and renal function | (these must be excluded first) |
Extra clues:
-
Low serum uric acid (uric acid is lost in urine)
-
Urine osmolality/serum osmolality ratio >2
-
Murray & Nadel's Textbook of Respiratory Medicine, 2-Volume Set
Causes of SIADH
CNS Disorders:
- Head trauma, meningitis, encephalitis
- Subarachnoid hemorrhage, stroke
- Brain tumors
- Pituitary surgery
Pulmonary Disorders:
- Pneumonia, tuberculosis
- Positive-pressure ventilation
- Small cell lung cancer (SCLC) - most important malignancy cause; SCLC accounts for ~75% of all SIADH cases (only ~10% of SCLC patients develop it, but SCLC is so common that it dominates the SIADH statistics)
Malignancies (ectopic ADH production):
- Small cell lung cancer (most common)
- Pancreatic, prostate, bladder cancers
Drugs (very common cause - memorize these):
- Carbamazepine (classic exam answer)
- SSRIs / SNRIs (antidepressants)
- NSAIDs
- Antipsychotics
- Morphine / opioids
- Cyclophosphamide (chemotherapy)
- MDMA / ecstasy (club drug - common in ER)
- Chlorpropamide (old sulfonylurea)
- Vincristine (chemo)
Other:
- Nausea, pain, surgery (physiologic stimuli that can be excessive)
- HIV/AIDS
Why Is Sodium Low in SIADH? (Mechanism)
- ADH is secreted inappropriately → V2 receptors activated in collecting duct
- AQP2 channels inserted → water reabsorbed from tubule → water enters blood
- Blood becomes diluted (too much water relative to solute)
- Serum osmolality falls → serum Na falls (hyponatremia)
- The expanded blood volume causes the kidneys to excrete some sodium (natriuresis) to try to "offload" volume → urine Na is HIGH (paradox - body is losing sodium despite low blood sodium)
- The normal feedback (low osmolality → suppress ADH) is broken in SIADH → ADH keeps secreting regardless
Symptoms of SIADH (= Symptoms of Hyponatremia)
Severity depends on how low sodium drops and how fast:
| Serum Na | Typical Symptoms |
|---|---|
| 130-135 mEq/L | Often asymptomatic; mild headache, difficulty concentrating |
| 120-130 mEq/L | Nausea, vomiting, confusion, irritability, gait disturbance, falls |
| <120 mEq/L | Lethargy, obtundation, severe confusion |
| <115 mEq/L | Seizures, coma, respiratory arrest, brain herniation - life-threatening |
Note: In one large series, only 27% of SIADH patients had symptoms even with a median sodium of 117 mEq/L - the brain adapts by extruding intracellular solutes (volume regulation). Chronic hyponatremia is better tolerated than acute.
Treatment of SIADH
Treatment is guided by symptom severity, not just sodium level.
Mild/Asymptomatic Hyponatremia:
- Fluid restriction (500-1000 mL/day) - first line
- Address underlying cause (stop offending drug, treat infection, treat tumor with chemo)
Moderate Symptoms (confusion, nausea, gait problems):
- Continue fluid restriction
- Add demeclocycline (900-1200 mg/day orally) - induces nephrogenic DI, blocks ADH action on kidneys. Onset 2-5 days, so not for emergencies. Watch for nephrotoxicity.
- Vaptans (tolvaptan oral or conivaptan IV) - V2 receptor antagonists, cause aquaresis (free water excretion without sodium loss)
Severe/Life-threatening (seizures, coma, Na <115 mEq/L):
- 3% Hypertonic NaCl - give 300 mL over 3-4 hours, often combined with a loop diuretic (furosemide)
- Why add a loop diuretic? Hypertonic saline alone can worsen volume overload; furosemide helps excrete water while keeping Na
- Target: raise Na by 1-2 mEq/L/hour initially until symptoms resolve
The Most Dangerous Complication of Treatment: Osmotic Demyelination Syndrome (ODS)
Formerly called Central Pontine Myelinolysis (CPM) - now ODS is the preferred term since demyelination can occur outside the pons too.
What is it?
When sodium has been chronically low, brain cells adapt by losing intracellular solutes to prevent brain swelling. If you then rapidly correct sodium, the blood becomes suddenly concentrated - water rushes OUT of brain cells - the cells shrink - and the myelin sheaths of axons in the pons are destroyed.
Result: A devastating neurological syndrome developing 2-4 days after sodium correction:
- Quadriplegia (paralysis of all 4 limbs)
- Dysarthria (slurred speech)
- Dysphagia (difficulty swallowing)
- Pseudobulbar palsy
- Altered mental status, locked-in syndrome
MRI finding: Symmetric T2 high signal in the central pons (sparing the corticospinal tracts)
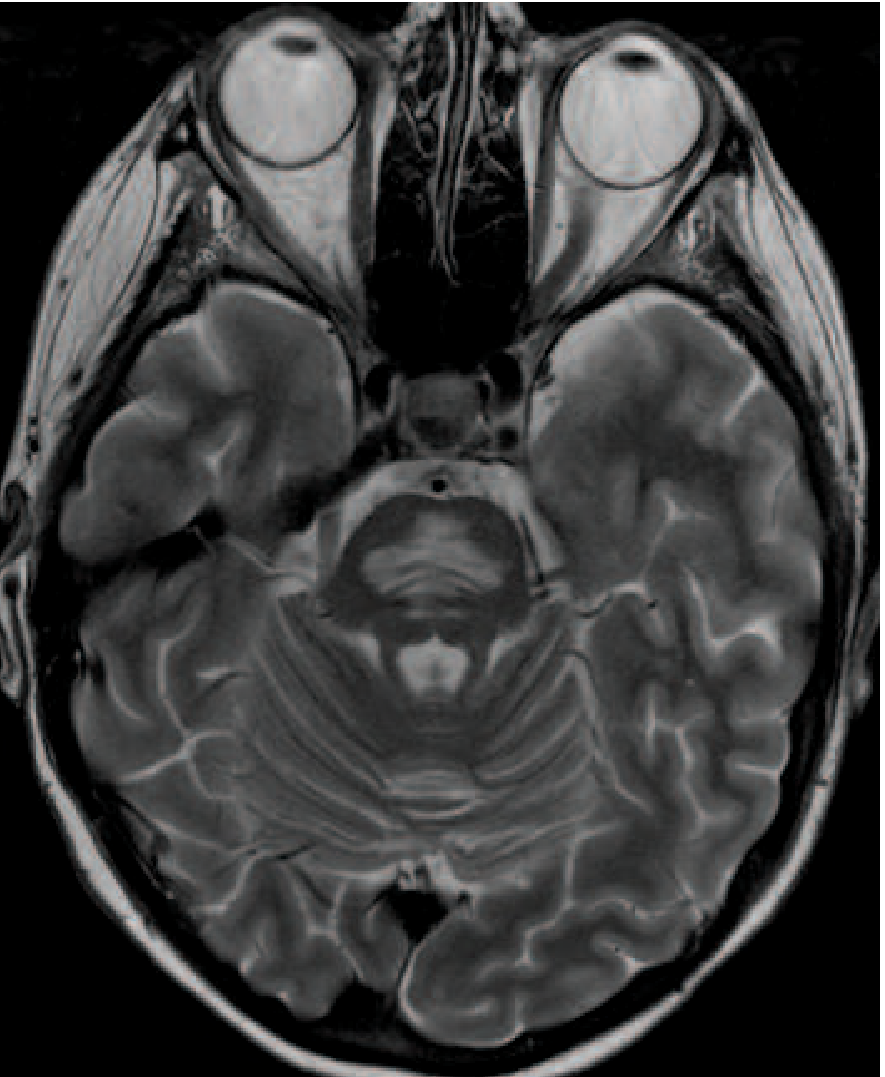
The safe rate of correction:
- Maximum: ≤8 mEq/L in 24 hours and ≤15 mEq/L in 48 hours
- Never exceed 20 mEq/L in the first 24 hours
- Check sodium levels frequently (every 2-4 hours) when actively treating
Key rule: Go slow. Too fast = ODS.
- Harrison's Principles of Internal Medicine, 22nd Ed.
PART 3: DI vs SIADH - The Master Comparison Table
| Feature | DI | SIADH |
|---|---|---|
| ADH activity | Absent/ineffective | Excessive |
| Serum Na | HIGH (hypernatremia) | LOW (hyponatremia) |
| Serum osmolality | HIGH (>295 mOsm/kg) | LOW (<275 mOsm/kg) |
| Urine osmolality | LOW (<300 mOsm/kg) | HIGH (>200 mOsm/kg, concentrated) |
| Urine volume | Massive (up to 20 L/day) | Low |
| Urine Na | Low | HIGH (>20 mEq/L) |
| Thirst | Extreme | Normal or absent |
| Volume status | Dehydrated (or normal) | Euvolemic |
| Key symptom | Polyuria + polydipsia | Hyponatremia symptoms |
| Treatment | Desmopressin (central) / thiazides (nephrogenic) | Fluid restriction / vaptans / hypertonic saline |
PART 4: Memory Mnemonics
SIADH causes - "PIN DOWN":
- Pulmonary disease (pneumonia, TB)
- Infection (meningitis, encephalitis)
- Neoplasm (SCLC, pancreatic ca)
- Drugs (carbamazepine, SSRIs, opioids)
- Operation/surgery (post-op stress)
- Whacking brain (head trauma, SAH)
- Not (normal) thyroid, adrenal (these must be normal for diagnosis)
DI vs SIADH urine/blood:
- DI = Dilute urine, high serum (D for Dry blood)
- SIADH = Concentrated urine, dilute serum (S for Soggy blood)
ODS correction rule: "8 in 24, 15 in 48" - never exceed these sodium rise rates.
Sources: Goldman-Cecil Medicine, International Ed. | Harrison's Principles of Internal Medicine, 22nd Ed. | Murray & Nadel's Textbook of Respiratory Medicine | Brenner and Rector's The Kidney, 2-Volume Set | Neuroanatomy through Clinical Cases, 3rd Ed. | Tietz Textbook of Laboratory Medicine, 7th Ed.
This is a shared conversation. Sign in to Orris to start your own chat.