Apoptosis – (AP 22) a. Definition of Apoptosis b. Enumerate in Detail about the Pathways of Apoptosis c. What are the Other Methods of Cell Death? 15 marks question for mbbs pathology University exam knruhs University
Reading File
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Finding Sources
Reading File
Finding Sources
Finding Sources
Reading File
Finding Sources
Reading File
Reading File
Loading Image
I now have all the content needed. Let me compile a comprehensive, exam-ready answer.
Apoptosis — AP 22 (15 Marks)
Source: Robbins & Kumar — Pathologic Basis of Disease / Basic Pathology
A. Definition of Apoptosis
Apoptosis (Greek: "falling off," as leaves fall from a tree) is a pathway of programmed cell death in which cells activate intrinsic enzymes that degrade their own nuclear DNA and nuclear and cytoplasmic proteins in a controlled, energy-dependent manner.
Key distinguishing features:
- The plasma membrane remains intact throughout, but is altered to display "eat-me" signals (e.g., phosphatidylserine flip to outer leaflet)
- The cell fragments into membrane-bound apoptotic bodies, which are rapidly phagocytosed by macrophages
- No inflammatory reaction is elicited because cellular contents do not leak into the extracellular space (contrast with necrosis)
- It occurs in both physiologic (normal development, immune homeostasis) and pathologic conditions (DNA damage, viral infections)
"Apoptosis is a pathway of cell death in which cells activate enzymes that degrade the cells' own nuclear DNA and nuclear and cytoplasmic proteins." — Robbins & Kumar Basic Pathology
Morphologic Features of Apoptosis
| Feature | Description |
|---|---|
| Cell shrinkage | Cell reduces in size; cytoplasm becomes dense |
| Chromatin condensation | Chromatin aggregates into dense crescents (pyknosis) |
| Nuclear fragmentation | Karyorrhexis — nucleus breaks into pieces |
| Apoptotic bodies | Membrane-bound fragments containing organelles/nuclear debris |
| Phagocytosis | Rapid uptake by neighboring cells/macrophages |
| No inflammation | Membrane intact → no leakage of contents |
B. Pathways of Apoptosis (Mechanisms in Detail)
Apoptosis is regulated by biochemical pathways that balance death- and survival-inducing signals, ultimately activating caspases — cysteine proteases that cleave proteins after aspartic acid residues.
There are two principal pathways, both converging on caspase activation:
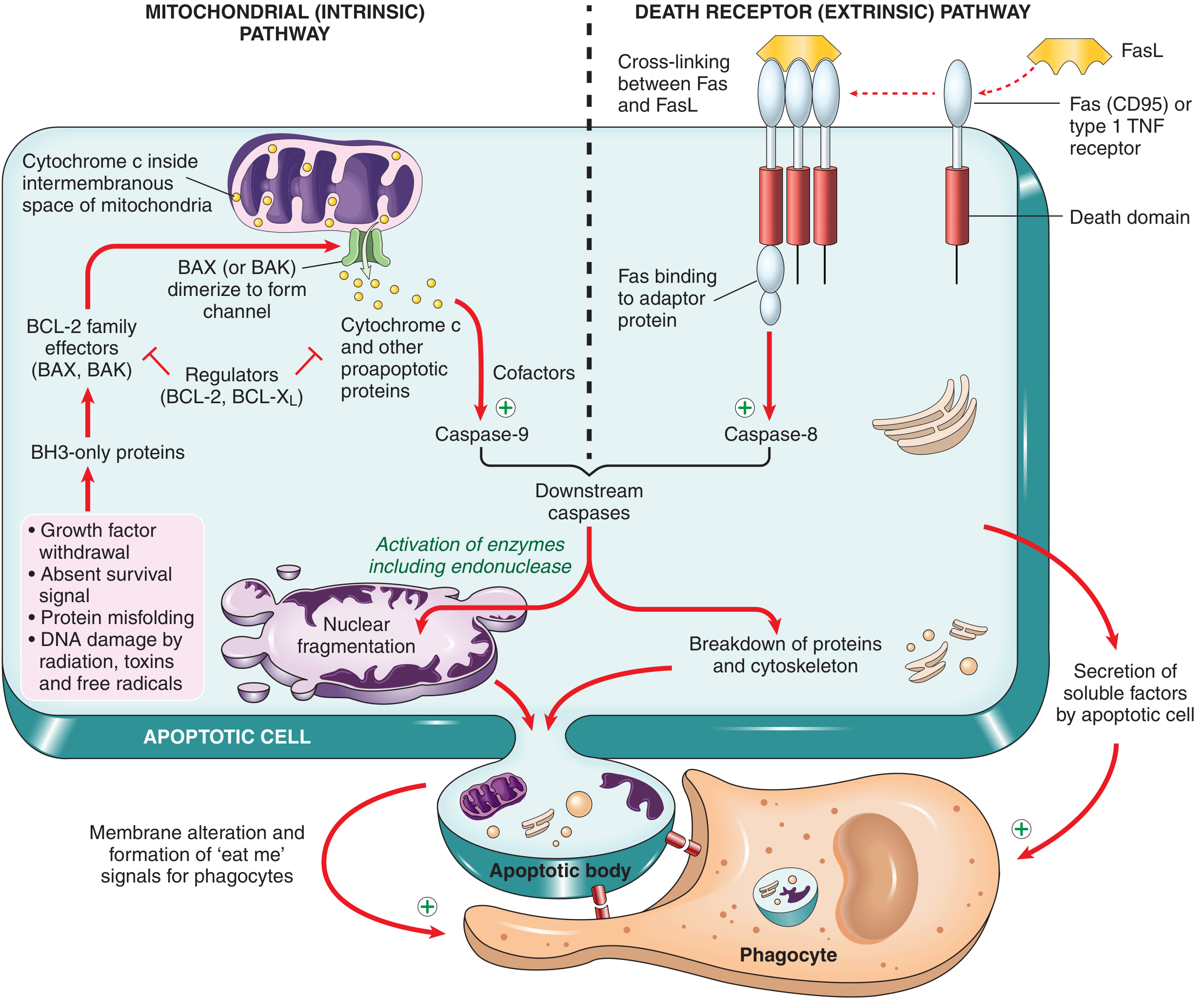
1. Mitochondrial (Intrinsic) Pathway
This is responsible for apoptosis in most physiologic and pathologic situations.
Triggers:
- Withdrawal of growth factors/survival signals
- DNA damage (radiation, toxins, free radicals)
- Accumulation of misfolded proteins (ER stress)
- Oxidative stress
Mechanism — Step by Step:
Step 1 — Sensor activation (BH3-only proteins):
When cells are deprived of survival signals or suffer DNA/protein damage, BH3-only proteins are activated. These are sensors that contain the third homology domain (BH3) of the BCL-2 family.
Step 2 — BCL-2 family balance shifts:
- Anti-apoptotic proteins: BCL-2 and BCL-X_L are produced in response to growth factors; they maintain mitochondrial membrane integrity by holding BAX/BAK in check.
- Pro-apoptotic proteins: BAX and BAK are two effectors that, when freed from BCL-2/BCL-X_L inhibition by BH3-only proteins, dimerize and insert into the outer mitochondrial membrane, forming channels.
Step 3 — Cytochrome c release:
BAX/BAK channels allow cytochrome c (and other pro-apoptotic mitochondrial proteins such as Smac/DIABLO) to leak from the intermembranous space into the cytosol.
Step 4 — Apoptosome formation:
Cytochrome c binds APAF-1 (apoptosis-activating factor-1) in the cytosol → forms a multimeric wheel-like complex called the apoptosome.
Step 5 — Initiator caspase activation:
The apoptosome recruits and activates caspase-9 (the initiator caspase of this pathway) by autocatalytic cleavage.
Step 6 — Executioner caspase cascade:
Active caspase-9 cleaves and activates executioner caspases (caspase-3, caspase-6), which then:
- Activate a DNase (CAD) that degrades nuclear DNA into oligonucleosomal fragments → "DNA ladder" on gel electrophoresis
- Proteolyze structural components of the nuclear matrix → nuclear fragmentation
- Break down cytoskeletal proteins → cell shrinkage and blebbing
Smac/DIABLO role: These mitochondrial proteins inhibit IAPs (Inhibitor of Apoptosis Proteins), thereby releasing the brake on caspase-3 and ensuring the death cascade proceeds.
2. Death Receptor (Extrinsic) Pathway
This pathway is initiated by signals from plasma membrane death receptors.
Death receptors: Members of the TNF receptor superfamily containing a conserved cytoplasmic "death domain" — e.g.:
- TNFR1 (Type 1 TNF receptor)
- Fas (CD95) — the best-characterized death receptor
Ligands:
- FasL (Fas ligand) — expressed on cytotoxic T lymphocytes (CTLs) and self-reactive T cells
- TNF-α
Mechanism — Step by Step:
Step 1 — Receptor trimerization:
FasL binds Fas → three or more Fas molecules cluster together on the cell surface.
Step 2 — FADD recruitment:
Clustered death domains recruit the adaptor protein FADD (Fas-Associated Death Domain protein).
Step 3 — DISC formation:
FADD recruits pro-caspase-8 (or caspase-10) → formation of DISC (Death-Inducing Signaling Complex).
Step 4 — Initiator caspase activation:
Multiple pro-caspase-8 molecules in the DISC undergo autocatalytic cleavage → active caspase-8.
Step 5 — Executioner caspase activation:
Active caspase-8 activates the same executioner caspase cascade (caspase-3, caspase-6), leading to the same end result as the intrinsic pathway.
Inhibition: FLIP (FLICE-inhibitory protein) — a decoy protein that binds pro-caspase-8 and blocks DISC formation. Some viruses produce FLIP to protect infected cells from Fas-mediated killing.
3. Convergence of Both Pathways — The Execution Phase
| Feature | Intrinsic | Extrinsic |
|---|---|---|
| Trigger | Intracellular stress, DNA damage | External death ligands (FasL, TNF) |
| Initiator caspase | Caspase-9 | Caspase-8/10 |
| Key complex | Apoptosome (Cyt c + APAF-1 + Casp-9) | DISC (Fas-FADD-Casp-8) |
| Executioner caspases | Caspase-3, -6, -7 | Caspase-3, -6, -7 |
| Key regulators | BCL-2 family, BH3-only proteins | FLIP, IAPs |
Both pathways converge on the execution caspases (caspase-3, -6) which bring about:
- Nuclear DNA fragmentation (by CAD endonuclease)
- Nuclear membrane dissolution
- Cytoskeletal breakdown
- Membrane blebbing → apoptotic bodies
- Phosphatidylserine exposure → "eat-me" signal for phagocytes
Cross-talk: In hepatocytes/pancreatic β-cells, caspase-8 (extrinsic) cleaves the BH3-only protein BID → truncated BID (tBID) activates the mitochondrial pathway, amplifying the apoptotic signal.
4. Role of p53 in Apoptosis
- DNA damage → stabilizes p53 (tumor suppressor protein)
- p53 upregulates BAX and BH3-only proteins (e.g., PUMA, NOXA)
- This tilts the BCL-2 balance toward pro-apoptotic proteins → intrinsic pathway activation
- Loss of p53 (e.e., mutation in cancer cells) allows cells with damaged DNA to survive → carcinogenesis
Summary Diagram of Intrinsic Pathway:
Growth factor withdrawal / DNA damage / Misfolded proteins
↓
BH3-only proteins activated (PUMA, NOXA, BIM)
↓
BCL-2/BCL-XL inhibited → BAX/BAK freed
↓
BAX/BAK dimerize → insert into outer mitochondrial membrane
↓
Cytochrome c + Smac/DIABLO released into cytosol
↓
Cytochrome c + APAF-1 → APOPTOSOME
↓
Caspase-9 activated (initiator)
↓
Caspase-3 / Caspase-6 activated (executioners)
↓
DNA fragmentation, nuclear collapse, apoptotic bodies
C. Other Methods of Cell Death
Beyond classical apoptosis and necrosis, several additional regulated cell death programs have been characterized:
1. Necroptosis ("Programmed Necrosis")
- Definition: A form of regulated cell death that morphologically resembles necrosis but is genetically programmed (like apoptosis)
- Triggers: Ligation of TNFR1, viral RNA/DNA sensors (e.g., RIG-I, DAI), and other ligands
- Mechanism: Activated by a kinase called RIPK3 (receptor-interacting protein kinase 3), which phosphorylates MLKL (mixed lineage kinase domain-like protein) → MLKL inserts into the plasma membrane → loss of membrane integrity → cell swelling and rupture
- Key feature: Occurs when caspases are inhibited (e.g., in viral infections where the virus blocks caspase-8)
- Significance: Releases DAMPs (damage-associated molecular patterns) → triggers inflammation; may contribute to ischemia-reperfusion injury, neurodegeneration, inflammatory bowel disease
2. Pyroptosis ("Fiery Death")
- Definition: A highly inflammatory form of programmed cell death associated with activation of caspase-1
- Triggers: Intracellular danger signals (e.g., cytosolic LPS, uric acid crystals, bacterial toxins) detected by NOD-like receptors (NLRs)
- Mechanism:
- Activated NLRs → assembly of the inflammasome (multiprotein complex)
- Inflammasome activates caspase-1 → cleaves pro-IL-1β and pro-IL-18 into active, secreted forms
- Caspase-1 also cleaves gasdermin D → N-terminal fragment inserts into the plasma membrane → pore formation → cell swelling and lysis
- Also activated by caspase-4/5 (human) in response to intracellular LPS
- Significance: Important in innate immune defense; contributes to the pathogenesis of gout, atherosclerosis, and septic shock
3. Ferroptosis ("Iron-dependent Death")
- Definition: A form of cell death driven by iron-dependent accumulation of oxidized lipids (lipid peroxidation) — morphologically and mechanistically distinct from apoptosis and necrosis
- Triggers: Depletion of glutathione peroxidase 4 (GPX4), which normally neutralizes lipid peroxides; excess intracellular iron
- Mechanism:
- Iron (via Fenton reaction) + polyunsaturated fatty acids → lipid peroxides accumulate
- GPX4 normally detoxifies these; when GPX4 is inhibited (e.g., by erastin/RSL3), peroxides accumulate → membrane damage → cell death
- Morphology: Shrunken mitochondria with increased membrane density; no nuclear fragmentation (distinguishes it from apoptosis)
- Significance: Implicated in neurodegeneration, acute kidney injury, ischemia-reperfusion injury, and cancer cell death
4. Autophagy-Associated Cell Death
- Definition: Autophagy is primarily a survival mechanism (degradation of damaged organelles); however, excessive autophagy can lead to cell death
- Mechanism: Cellular contents are engulfed in autophagosomes → fuse with lysosomes → contents degraded; when overwhelming, leads to cell death
- Significance: Implicated in neurodegeneration, cancer, starvation-induced cell death
Summary Table — Cell Death Mechanisms
| Feature | Apoptosis | Necrosis | Necroptosis | Pyroptosis | Ferroptosis |
|---|---|---|---|---|---|
| Programmed? | Yes | No | Yes | Yes | Yes |
| Membrane intact? | Yes | No | No | No | No |
| Inflammation? | No | Yes | Yes | Yes (major) | Yes |
| Caspase involved? | Yes (3, 8, 9) | No | No (RIPK3/MLKL) | Yes (Casp-1) | No |
| DNA fragmentation? | Yes | Non-specific | No | No | No |
| Key mediator | BCL-2/Cyt c/Casp | Non-specific damage | RIPK3/MLKL | Gasdermin D | GPX4/lipid peroxides |
| Example | Embryogenesis, immune cells | MI, ischemia | Viral infection | Gout, sepsis | Neurodegeneration |
Key Points for Exam (KNRUHS)
- Apoptosis = programmed, non-inflammatory, energy-dependent cell death; membrane remains intact
- Two major pathways: Intrinsic (mitochondrial/BCL-2/caspase-9) and Extrinsic (death receptor/Fas-FADD/caspase-8)
- Both converge on caspase-3/6 (executioners) → DNA ladder + apoptotic bodies
- p53 links DNA damage to apoptosis via BH3-only proteins
- Other cell death mechanisms: Necroptosis (RIPK3/MLKL), Pyroptosis (caspase-1/gasdermin D), Ferroptosis (GPX4/lipid peroxides)
Sources: Robbins & Kumar Basic Pathology (10th ed.) p. 24–45; Robbins, Cotran & Kumar Pathologic Basis of Disease (10th ed.) p. 38–61
This is a shared conversation. Sign in to Orris to start your own chat.