Nicotinic and muscarinic difference
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Finding Sources
Finding Sources
Reading File
Finding Sources
Reading File
Reading File
Loading Image
Here is a thorough comparison of nicotinic and muscarinic receptors, drawn from Lippincott Illustrated Reviews: Pharmacology, Katzung's Basic and Clinical Pharmacology, and Barash's Clinical Anesthesia.
Nicotinic vs. Muscarinic Receptors
Both receptor types are cholinoceptors - they both respond to acetylcholine (ACh) as their endogenous ligand, but they differ fundamentally in structure, location, signaling mechanism, and clinical pharmacology.
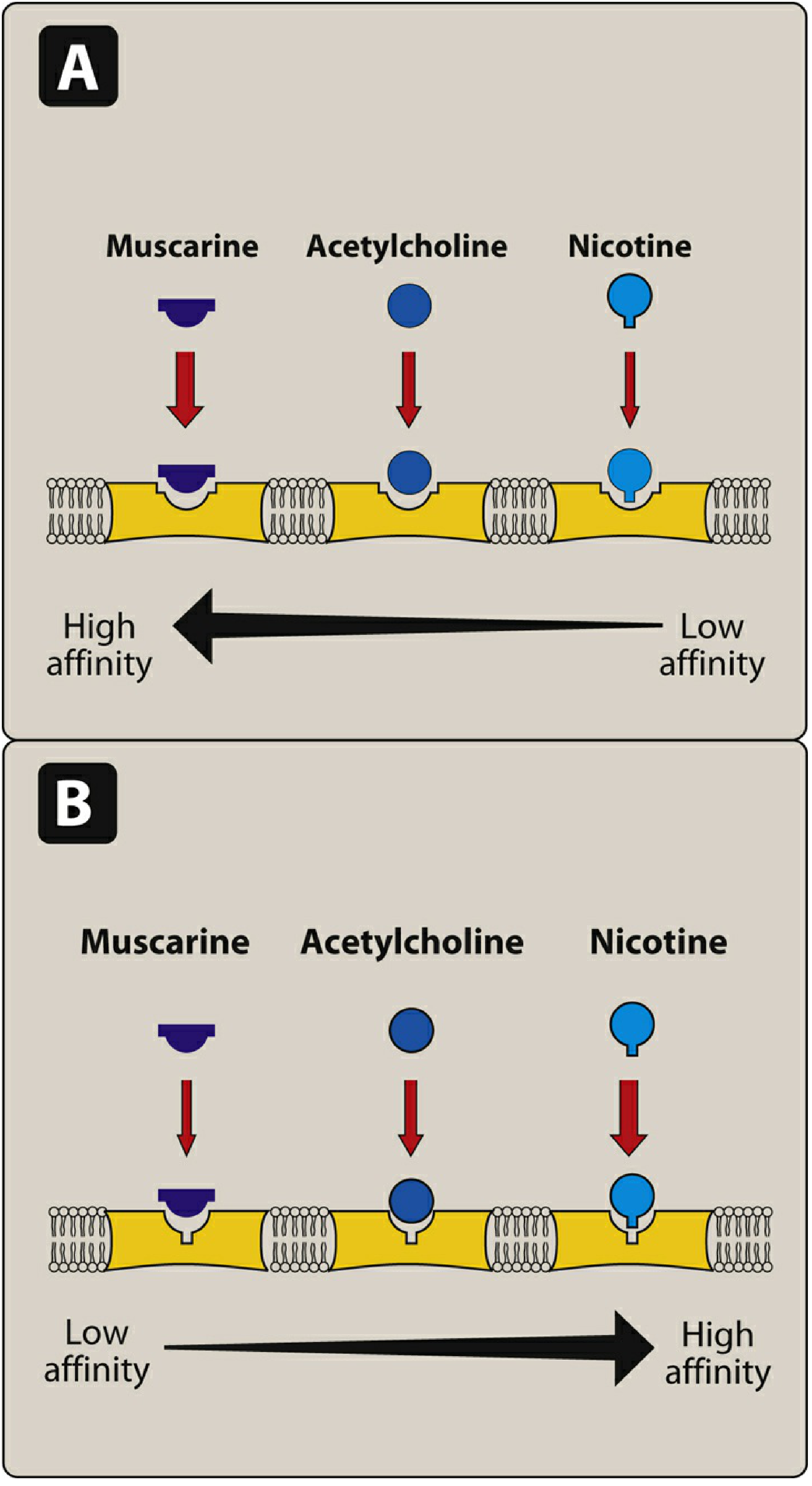
1. Receptor Type and Mechanism
| Feature | Nicotinic | Muscarinic |
|---|---|---|
| Receptor class | Ionotropic (ligand-gated ion channel) | Metabotropic (G protein-coupled receptor, GPCR) |
| Signal onset | Fast (milliseconds) | Slow (seconds) |
| Ion movement | Na+ in, K+ out → depolarization | Via second messengers (IP3/DAG, cAMP) |
| Subunits | 5 subunits (2α + β + γ + δ); 2 ACh molecules must bind | Single polypeptide with 7 transmembrane domains |
Nicotinic: Binding of 2 ACh molecules causes a conformational change opening the central ion channel, allowing Na+ influx and K+ efflux, producing rapid depolarization. This is the fastest known receptor mechanism in neurotransmission. (Lippincott Pharmacology, p. 150)
Muscarinic: All 5 subtypes (M1-M5) are GPCRs. They signal through second messenger cascades:
- M1, M3, M5 couple to Gq → activate phospholipase C → IP3 + DAG → ↑ intracellular Ca²+ and protein kinase C activation
- M2, M4 couple to Gi/o → inhibit adenylyl cyclase → ↓ cAMP; also open K+ channels (hyperpolarization in cardiac cells) (Katzung, p. 174; Lippincott, p. 149)
2. Location
| Location | Nicotinic (NM) | Nicotinic (NN) | Muscarinic |
|---|---|---|---|
| Neuromuscular junction (skeletal) | ✓ | - | - |
| Autonomic ganglia (both SNS + PNS) | - | ✓ | - |
| Adrenal medulla | - | ✓ | - |
| CNS | + | ✓ | ✓ |
| Cardiac muscle | - | - | ✓ (M2) |
| Smooth muscle | - | - | ✓ (M2, M3) |
| Exocrine glands | - | - | ✓ (M3) |
| Gastric parietal cells | - | - | ✓ (M1) |
NM = neuromuscular nicotinic; NN = neuronal/ganglionic nicotinic.
Ganglionic nicotinic receptors differ from NMJ receptors: ganglionic receptors are blocked by mecamylamine, while NMJ receptors are blocked by neuromuscular blockers like atracurium or vecuronium. (Lippincott, p. 150)
3. Muscarinic Subtypes
| Subtype | G-protein | Location | Effect |
|---|---|---|---|
| M1 | Gq | Gastric parietal cells, neurons, CNS | ↑ gastric acid secretion; CNS excitation |
| M2 | Gi | Heart (SA node, AV node), smooth muscle | ↓ HR, ↓ contractility, ↑ K+ conductance |
| M3 | Gq | Smooth muscle, glands, lungs, bladder, eye | Contraction, gland secretion, bronchoconstriction, miosis |
| M4 | Gi | CNS | Modulation of dopamine |
| M5 | Gq | CNS | Modulation of dopamine reward |
Only M1, M2, and M3 have been well characterized pharmacologically. (Lippincott, p. 149; Barash Clinical Anesthesia, p. 1049)
4. Selective Agonists and Antagonists
| Agent | Action | Target |
|---|---|---|
| Nicotine (low dose) | Stimulates | Nicotinic (all) |
| Nicotine (high dose) | Blocks | Nicotinic (ganglionic) |
| Muscarine | Stimulates | Muscarinic |
| Atropine | Blocks | Muscarinic (all subtypes) |
| Mecamylamine | Blocks | Nicotinic ganglionic (NN) |
| Atracurium / vecuronium | Blocks | Nicotinic NMJ (NM) |
| Bethanechol, pilocarpine | Stimulate | Muscarinic (preferential) |
Atropine reverses muscarinic effects (e.g., bronchorrhea, bradycardia) in organophosphate poisoning but does NOT reverse the muscle weakness caused by nicotinic receptor activation at the NMJ. (Barash, p. 1049; Morgan & Mikhail's Clinical Anesthesiology)
5. Physiological Effects Compared
Muscarinic stimulation (parasympathomimetic - SLUDGE/DUMBELS):
- Heart: bradycardia, ↓ contractility (M2)
- Eye: miosis, accommodation (M3)
- GI: hypermotility, ↑ secretions (M2, M3)
- Lungs: bronchoconstriction, ↑ secretions (M3)
- Bladder: detrusor contraction, ↑ micturition (M3)
- Glands: salivation, lacrimation, sweating (M3)
Nicotinic stimulation:
- Skeletal muscle: contraction (NMJ, NM)
- Autonomic ganglia: stimulates both SNS + PNS postganglionic neurons (NN)
- Adrenal medulla: releases epinephrine/NE (NN)
- At high doses: ganglionic blockade → hypotension + weakness
6. Clinical Pearls
- Organophosphate/nerve agent poisoning: Both receptors are overwhelmed. Atropine reverses muscarinic effects; pralidoxime (2-PAM) must be given to reverse the nicotinic (NMJ weakness) effects by reactivating acetylcholinesterase.
- Anticholinesterases (e.g., neostigmine) increase ACh everywhere - so muscarinic side effects (bradycardia, salivation) occur at lower concentrations than the desired nicotinic effect (reversing neuromuscular blockade). That is why atropine or glycopyrrolate is co-administered when reversing NMJ blockade.
- Succinylcholine acts as a depolarizing NMJ blocker at nicotinic NM receptors.
Sources:
- Lippincott Illustrated Reviews: Pharmacology, pp. 147-150
- Katzung's Basic and Clinical Pharmacology (16th ed.), p. 174
- Barash, Cullen & Stoelting's Clinical Anesthesia (9th ed.), pp. 1049-1050
- Basic Medical Biochemistry (6th ed.), pp. 322-323
Propofol
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Finding Sources
Reading File
Reading File
Reading File
Loading Image
Here is a comprehensive overview of propofol drawn from Barash's Clinical Anesthesia (9th ed.), Goodman & Gilman's Pharmacological Basis of Therapeutics, and Miller's Anesthesia (10th ed.).
Propofol
Chemical name: 2,6-diisopropylphenol
Formulation: 1% propofol in 10% soybean oil, 2.25% glycerol, 1.2% egg phospholipid emulsifier - the familiar milky-white lipid emulsion. EDTA is added as an antimicrobial. Stored at room temperature.
1. Mechanism of Action
Propofol produces unconsciousness primarily by enhancing GABA-A receptor-mediated inhibition. It binds postsynaptically to GABA-A receptors on cortical pyramidal neurons, potentiating chloride influx and hyperpolarizing the cell. This enhanced GABAergic inhibition counteracts the ascending arousal inputs (from locus ceruleus, hypothalamus, midbrain, basal forebrain) that keep the cortex awake.
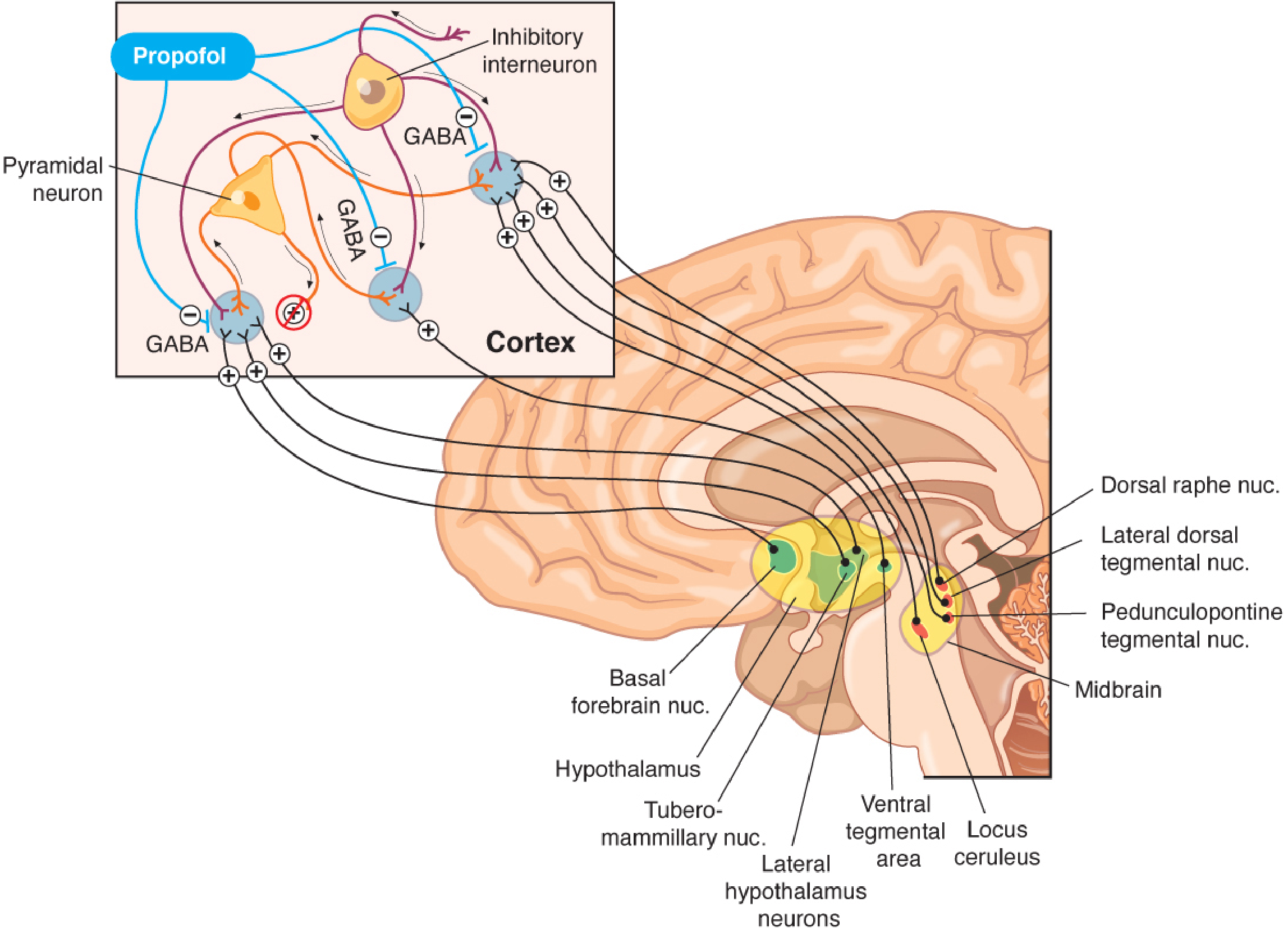
Additional (minor) mechanisms include:
- Inhibition of NMDA receptors
- Modulation of sodium and calcium channels
- Attenuation of excitotoxic glutamate pathways
2. Pharmacokinetics
| Parameter | Details |
|---|---|
| Onset | ~30-40 seconds (rapid, due to high lipid solubility) |
| Distribution t½ | 1-8 minutes (three-compartment model) |
| Elimination t½ | Context-sensitive; ~10 min after <3 hr infusion, <40 min after 8 hr infusion |
| Metabolism | Primarily hepatic (conjugation to inactive glucuronide/sulfate metabolites), plus significant extrahepatic metabolism (kidneys + lungs account for ~30%) |
| Clearance | 20-30 mL/kg/min - exceeds hepatic blood flow, hence extrahepatic routes are significant |
| Excretion | Renal (inactive metabolites); <3% excreted unchanged |
| Effect of liver/renal disease | Pharmacokinetics not significantly altered due to extrahepatic metabolism |
The context-sensitive half-time of propofol increases only modestly with prolonged infusions, making it suitable for total intravenous anesthesia (TIVA). (Goodman & Gilman, Fig. 24-3)
3. CNS Effects
| Effect | Details |
|---|---|
| Sedation → Unconsciousness | Dose-dependent; induction at ~3 mcg/mL plasma concentration |
| Paradoxical excitation | At intermediate doses: disinhibition, involuntary movement |
| EEG | Low dose: ↑ beta activity; induction: ↑ alpha/delta, ↓ beta (resembles deep non-REM sleep); high dose: burst suppression; very high dose: isoelectric EEG |
| Burst suppression | Achieved at ~8 mcg/mL; used for neuroprotection before aneurysm clipping |
| Anticonvulsant | Generally anticonvulsant; used to treat status epilepticus |
| Neuroprotection | ↓ CMRO2, ↓ CBF, ↓ ICP; free radical scavenger; anti-inflammatory (↓ TNF-α) |
| Antiemetic | Yes - even at sub-sedative doses (unique property) |
Note: Propofol shortens seizure duration and is therefore NOT the agent of choice for ECT.
The loss of consciousness from propofol can be partially reversed by physostigmine (a cholinomimetic), suggesting a cholinergic arousal component.
4. Cardiovascular Effects
- Significant decrease in systolic and diastolic blood pressure - the most clinically important hemodynamic effect
- Mechanism: ↓ cardiac output + ↓ stroke volume + ↓ systemic vascular resistance (SVR)
- Direct arterial vasodilation + venodilation (↓ sympathetic tone)
- Blunted baroreceptor reflex - expected reflex tachycardia is diminished
- May suppress supraventricular tachycardia (SVT)
- No expected compensatory rise in HR despite hypotension - clinically important in hypovolemic or cardiovascular-compromised patients
5. Respiratory Effects
- Dose-dependent respiratory depression
- Apnea common with induction doses
- At maintenance doses: ↓ tidal volume, ↑ respiratory rate
- Blunted hypoxic and hypercarbic ventilatory responses
- Potent bronchodilator (direct effect on intracellular calcium) - preferred induction agent in asthma
6. Clinical Uses
| Use | Dose |
|---|---|
| Induction of general anesthesia | 1-2.5 mg/kg IV (healthy adult) |
| TIVA maintenance | 100-200 mcg/kg/min infusion |
| Procedural sedation / conscious sedation | 25-75 mcg/kg/min |
| ICU sedation | Lower infusion rates; max 4 mg/kg/h (FDA limit) |
| PONV prophylaxis | Sub-hypnotic dose (10-20 mg bolus) |
| Status epilepticus | IV infusion |
Dose adjustments:
- Elderly: reduced dose (decreased cardiac output + clearance; prolonged effect)
- Children: increased mg/kg dose (larger volume of distribution, faster clearance)
- Morbidly obese: use lean body weight for dosing
- Chronic alcohol users: increased dose requirement
Propofol is safe in malignant hyperthermia (does not trigger MH).
7. Side Effects
| Side Effect | Details |
|---|---|
| Pain on injection | 60-70% of patients with peripheral IV (especially hand veins). Reduced by: IV lidocaine pretreatment with tourniquet (modified Bier block), using antecubital vein, adding lidocaine to propofol, or opioid pretreatment |
| Propofol Infusion Syndrome (PRIS) | Rare but potentially fatal (mortality ~50%). Features: metabolic acidosis, hyperkalemia, hyperlipidemia, rhabdomyolysis, hepatomegaly, renal failure, ECG changes, arrhythmias, cardiac failure. Mechanism: probable mitochondrial toxicity + inhibition of fatty acid oxidation. Risk: high doses (>4 mg/kg/h), prolonged infusion, young patients, head-injured patients |
| Hypotension | Especially prominent at induction |
| Hypertriglyceridemia | From lipid emulsion vehicle |
| Green urine | Rare but benign; extrahepatic metabolites; occurs after prolonged infusion |
| Abuse potential | 18% of US academic institutions report propofol diversion/abuse, primarily among anesthesia providers; associated with euphoria on emergence |
8. Contraindications / Cautions
- Egg/soy allergy: formulation contains egg phospholipid and soybean oil - caution warranted (though anaphylaxis risk is low)
- Severe cardiovascular compromise/hypovolemia: pronounced hypotension
- Pediatric ICU sedation (prolonged): high risk of PRIS - not recommended for long-term sedation in children
- ECT: shortens seizure duration - avoid
- Fospropofol (prodrug) is the only scheduled substance; propofol itself is not federally controlled but abuse is well documented
Sources:
- Barash, Cullen & Stoelting's Clinical Anesthesia (9th ed.), pp. 1473-1480
- Goodman & Gilman's Pharmacological Basis of Therapeutics, pp. 1066-1077
- Miller's Anesthesia (10th ed.), pp. 976-979
This is a shared conversation. Sign in to Orris to start your own chat.