Detailed explanation of Hemolytic anemia for PATHOLOGY
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Loading Image
Loading Image
I now have all the content needed to write a comprehensive pathology explanation. Let me compile the full answer.
Hemolytic Anemia - Pathology (Robbins & Kumar)
Definition and Overview
Hemolytic anemias are a diverse group of disorders sharing one common feature: accelerated red cell destruction with shortening of the normal 120-day red cell lifespan. The resulting low tissue O₂ triggers erythropoietin release from the kidney, causing compensatory marrow erythroid hyperplasia and peripheral reticulocytosis. In severe cases, the erythropoietic drive can be so pronounced that extramedullary hematopoiesis develops in the liver, spleen, and lymph nodes.
Classification
By Defect Location
| Type | Mechanism | Examples |
|---|---|---|
| Intrinsic (intracorpuscular) | Defect within the red cell itself | Hereditary spherocytosis, sickle cell, thalassemia, G6PD deficiency, PNH |
| Extrinsic (extracorpuscular) | External forces/agents attack normal red cells | Autoimmune hemolytic anemia, mechanical hemolysis, malaria |
By Site of Hemolysis (clinically most useful)
Extravascular Hemolysis
Caused by defects that increase destruction of red cells by phagocytes, especially in the spleen. The spleen's splenic sinusoids require extreme red cell deformability to traverse. Any reduction in deformability causes red cells to become "stuck" and phagocytosed by resident splenic macrophages.
Features specific to extravascular hemolysis:
- Hyperbilirubinemia and jaundice - from hemoglobin degradation in macrophages
- Splenomegaly - due to work hyperplasia of phagocytes
- Pigment gallstones (cholelithiasis) - if long-standing
Intravascular Hemolysis
Injuries so severe that red cells burst within the circulation - from mechanical forces (e.g., defective heart valve turbulence), complement fixation, clostridial toxins, or heat.
Features specific to intravascular hemolysis:
- Hemoglobinemia - free Hb in plasma
- Hemoglobinuria - Hb passes into urine (small enough to cross renal filter)
- Hemosiderinuria - partially reabsorbed Hb processed to hemosiderin, lost when renal tubular cells are sloughed
- Iron deficiency - iron is lost in urine (unlike extravascular, where macrophage recycling is efficient)
Common to BOTH types:
- Decreased serum haptoglobin - macrophages "regurgitate" enough Hb during red cell consumption that haptoglobin (which binds free Hb and is then cleared) falls even in purely extravascular hemolysis
Specific Types of Hemolytic Anemia
1. Hereditary Spherocytosis
Genetics: Usually autosomal dominant; a more severe autosomal recessive form exists.
Pathogenesis:
Caused by inherited defects in the membrane skeleton - a network of proteins that stabilize the lipid bilayer.
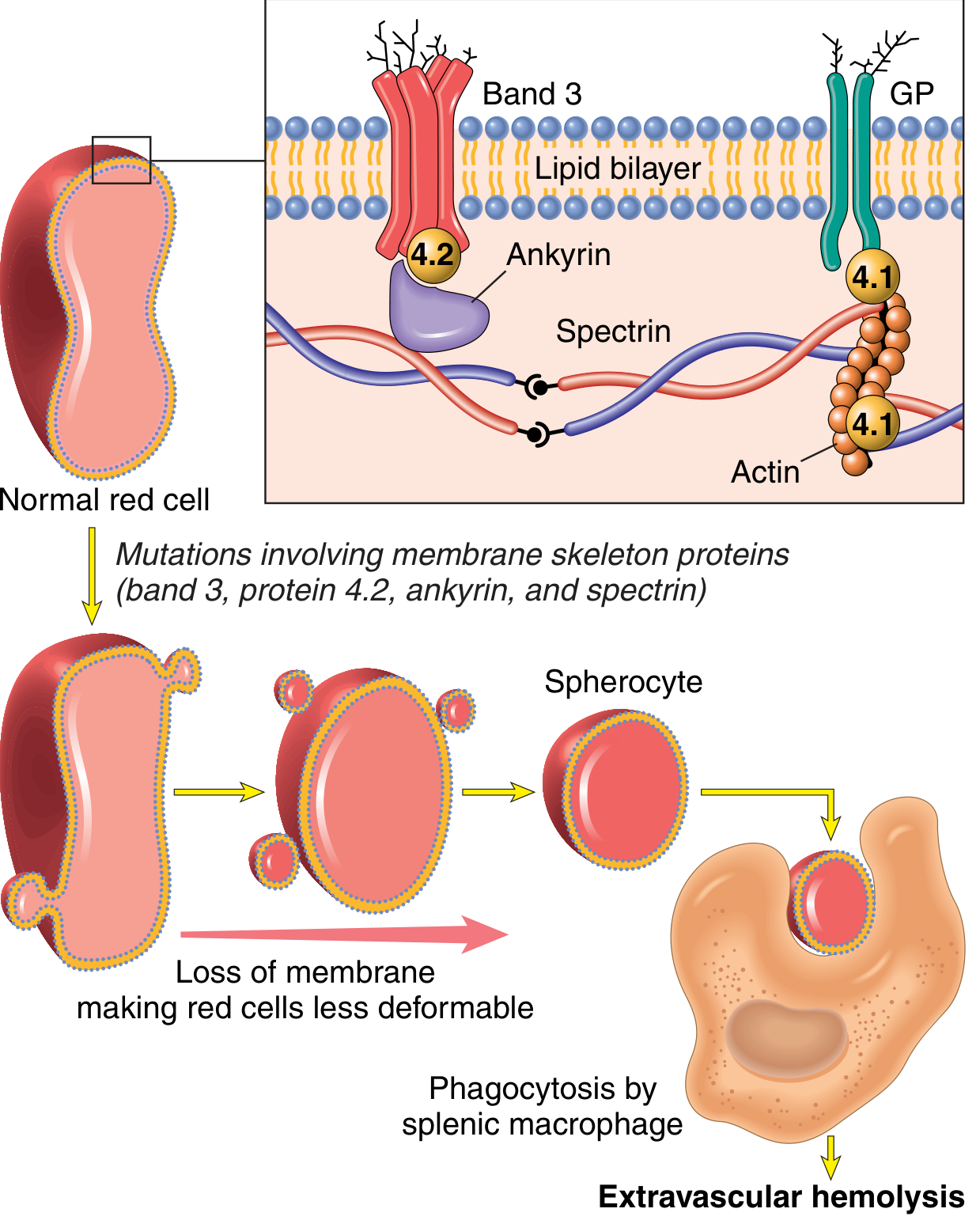
FIG. 10.1 - Normal red cell membrane skeleton (spectrin, ankyrin, band 4.1, band 4.2, band 3, glycophorin). Mutations weaken membrane skeleton-membrane protein interactions → vesicle shedding → spherocytes → trapped in splenic cords → phagocytosed by macrophages.
- Spectrin is the major skeleton protein (long, flexible heterodimer)
- Spectrin connects to intrinsic membrane proteins band 3 and glycophorin via ankyrin and band 4.1
- Mutations in spectrin, ankyrin, band 4.2, or band 3 weaken these interactions
- Result: Red cells shed membrane vesicles as they age → surface area-to-volume ratio decreases → spherocytes form
- Spherocytes are non-deformable → sequestered and destroyed in splenic cords (extravascular hemolysis)
- Splenectomy corrects the anemia (spherocytes persist but destruction stops)
Morphology:
- Peripheral smear: spherocytes are dark red, lack central pallor
- Splenomegaly is more prominent here than in any other hemolytic anemia (weight 500-1000 g; normal 150-200 g) - marked congestion of splenic cords, increased macrophages
- Cholelithiasis in 40-50% of patients
Lab: Osmotic fragility test - spherocytes lyse at lower osmolarity than normal RBCs.
2. Sickle Cell Anemia
Genetics: Autosomal recessive; most common familial hemolytic anemia. ~8% of African Americans are heterozygous carriers; ~1 in 600 have sickle cell anemia. HbS is protective against P. falciparum malaria.
Pathogenesis:
- Caused by a single amino acid substitution in β-globin: glutamate → valine at position 6
- On deoxygenation, HbS molecules polymerize via intermolecular contacts at the abnormal valine residue
- Polymers distort the red cell into an elongated crescentic (sickle) shape
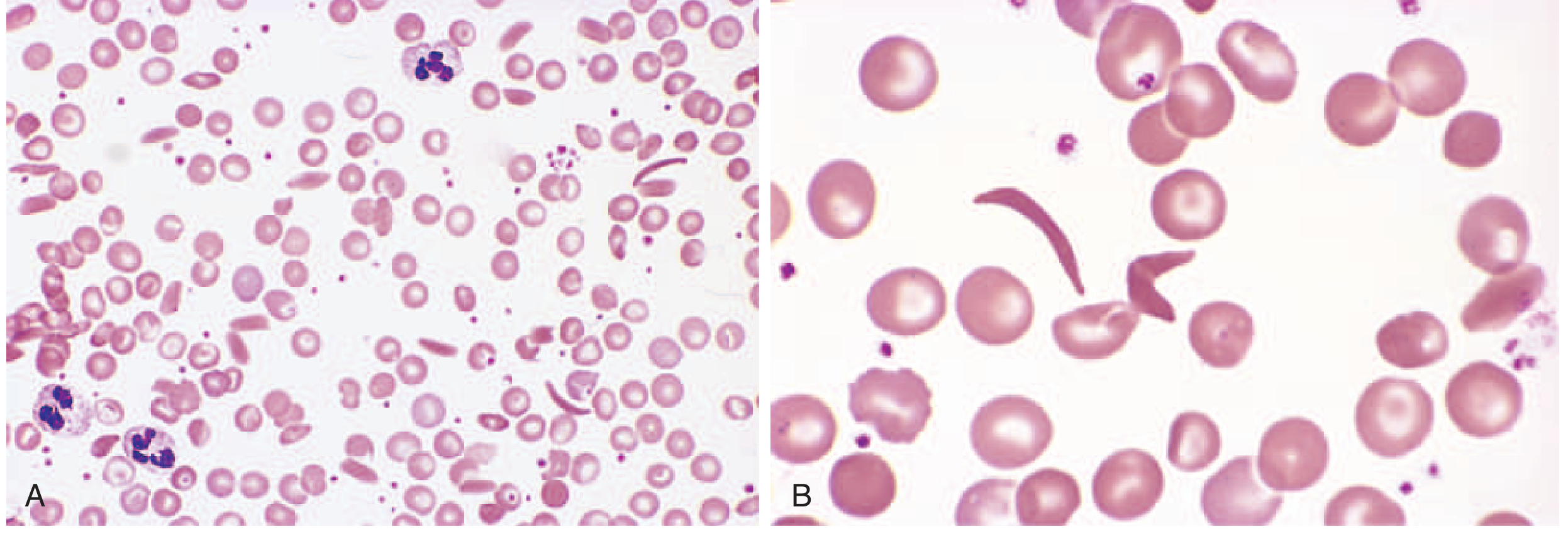
FIG. 10.3 - (A) Low-power view showing sickle cells and target cells. (B) High-power view showing elongated, crescent-shaped sickle cells.
Three key factors determining sickling:
- Intracellular non-HbS hemoglobin levels:
- HbA (in heterozygotes, ~40% HbS) greatly retards HbS polymerization → sickle cell trait (little tendency to sickle in vivo)
- HbF interacts weakly with HbS → newborns protected until HbF falls to adult levels (~5-6 months)
- HbC (lysine at position 6 instead of glutamate) - found in similar populations; in compound heterozygotes (HbSC disease), results in milder disease than homozygous HbSS
- Intracellular Hb concentration: increased MCHC (caused by dehydration) accelerates sickling
- Intracellular pH: low pH decreases O₂ affinity of Hb → more deoxygenation → more sickling
Consequences of sickling:
- Sickling is initially reversible on reoxygenation
- Repeated membrane distortion → calcium influx → loss of K⁺ and H₂O → membrane damage → irreversibly sickled cells that are prone to hemolysis
- Sickled cells obstruct microvasculature → ischemia/infarction in multiple organs
Morphology / Clinical Consequences:
| Feature | Mechanism |
|---|---|
| Chronic hemolytic anemia | Splenic trapping + intravascular lysis |
| Vaso-occlusive crises (pain crises) | Sickled cells obstruct small vessels |
| Autosplenectomy | Repeated splenic infarcts → fibrotic shrunken spleen |
| Increased infection risk | Impaired splenic function; also complement dysregulation |
| Acute chest syndrome | Vaso-occlusion + infarction in pulmonary vasculature |
| Stroke | Cerebral vessel occlusion |
| Bone marrow hyperplasia | Compensatory erythropoiesis → "crew-cut" skull on X-ray |
| Aplastic crisis | Parvovirus B19 infects erythroid progenitors |
| Leg ulcers | Skin ischemia |
| Priapism | Penile vascular occlusion |
3. Thalassemia
Definition: Inherited disorders caused by mutations that decrease synthesis of α-globin or β-globin, creating:
- Deficiency of hemoglobin (hypochromic, microcytic anemia)
- Intracellular precipitates of excess unpaired globin chains → red cell damage and hemolysis
Common in Mediterranean, African, and Asian regions (malaria-endemic areas - protective selection).
Genetics: Autosomal codominant.
- β-globin: single gene on chromosome 11
- α-globin: two tandem genes on chromosome 16
β-Thalassemia
| Syndrome | Genotype | Features |
|---|---|---|
| β-Thalassemia major | Homozygous (β⁰/β⁰ or β⁺/β⁺) | Severe anemia, regular transfusions required |
| β-Thalassemia intermedia | Variable | Moderate anemia, transfusions not required |
| β-Thalassemia minor (trait) | Heterozygous (β⁺/β) | Asymptomatic or mild; red cell changes seen |
Molecular cause: Mainly point mutations affecting transcription, splicing, or translation of β-globin mRNA.
- β⁰: complete absence of β-globin
- β⁺: reduced (but not absent) β-globin production
Pathogenesis of β-Thalassemia Major:
- Absent/markedly reduced β-globin → excess free α-chains that precipitate and form toxic inclusions in developing erythroid cells
- α-chain inclusions cause intravascular and intramedullary hemolysis (most red cell precursors die in the bone marrow - "ineffective erythropoiesis")
- Severe anemia → massive erythropoietin release → extreme marrow hyperplasia → cortical thinning of bone, facial deformity ("chipmunk facies"), pathological fractures
- Extramedullary hematopoiesis → hepatosplenomegaly
- Repeated blood transfusions → hemosiderosis (iron overload) → damage to heart, liver, endocrine organs (the major cause of death)
α-Thalassemia
| Syndrome | Missing Genes | Features |
|---|---|---|
| Silent carrier | 1 gene deleted (−/α, α/α) | Asymptomatic |
| α-Thalassemia trait | 2 genes deleted | Mild microcytic anemia |
| HbH disease | 3 genes deleted (−/−, −/α) | Moderately severe; HbH = β₄ tetramers |
| Hydrops fetalis | 4 genes deleted (−/−, −/−) | Hb Bart's = γ₄ tetramers; fatal in utero or at birth |
Molecular cause: Mainly gene deletions (unlike β-thalassemia which is mainly point mutations).
- With 3 deletions: excess β-chains form HbH (β₄) - soluble but unstable, precipitates → hemolysis
- With 4 deletions: excess γ-chains form Hb Bart's (γ₄) - very high O₂ affinity, delivers no O₂ to tissues → hydrops fetalis
4. Glucose-6-Phosphate Dehydrogenase (G6PD) Deficiency
Genetics: X-linked; most common inherited red cell enzyme deficiency worldwide.
Pathogenesis:
- G6PD is the rate-limiting enzyme of the hexose monophosphate shunt, generating NADPH
- NADPH maintains glutathione in reduced form → protects Hb and red cell membranes from oxidative damage
- G6PD-deficient cells cannot detoxify oxidants → Hb oxidized → precipitates as Heinz bodies (denatured globin)
- Macrophages "pluck out" Heinz bodies from red cells → "bite cells" on peripheral smear → these damaged cells are rapidly destroyed in the spleen
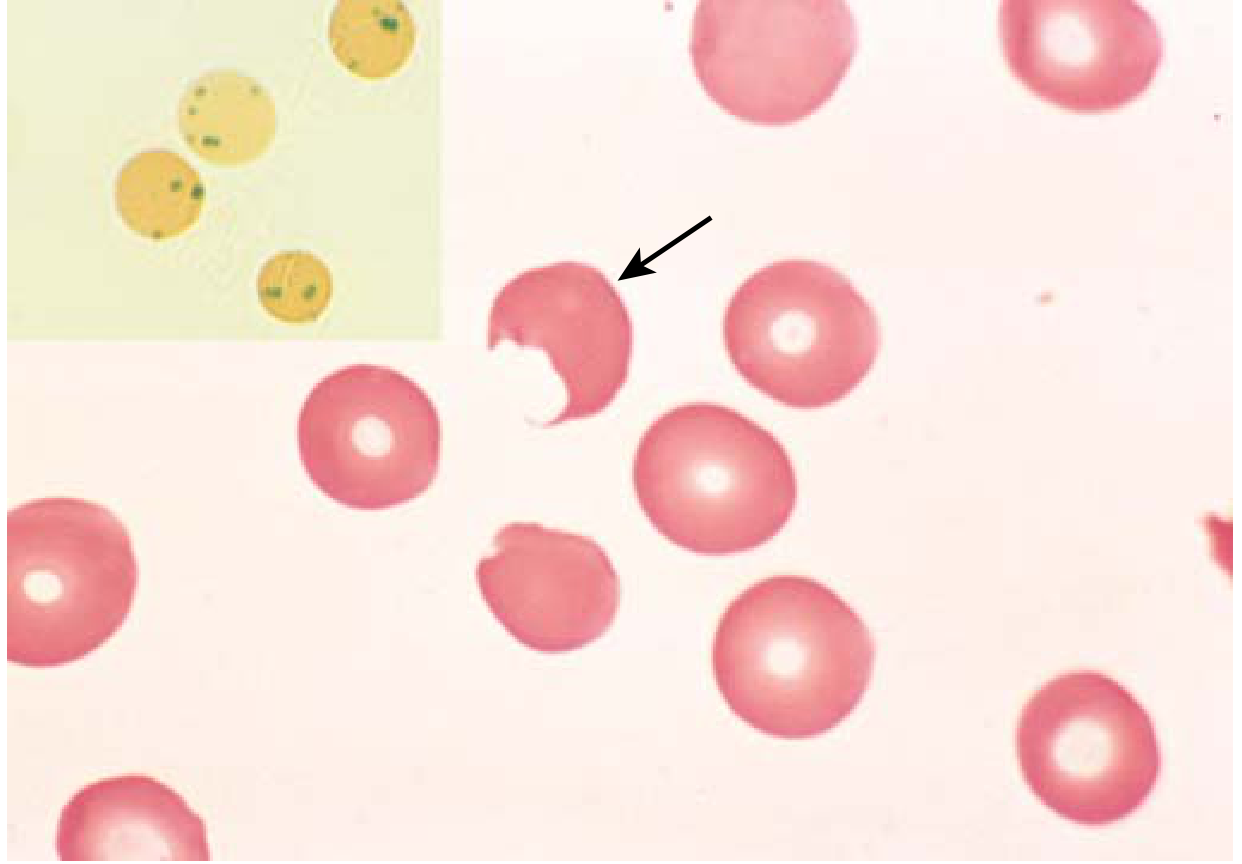
FIG. 10.6 - G6PD deficiency after oxidant exposure. Inset (supravital stain): Heinz bodies (denatured globin precipitates). Main: "bite cell" (arrow) where Heinz body has been plucked out by a splenic macrophage.
Triggers: Oxidant drugs (primaquine, dapsone, chloroquine), infections, fava beans
Important point: In the common African variant (G6PD A-), only older red cells are susceptible (shorter half-life enzyme) → hemolysis is self-limited even if drug exposure continues, because the marrow replaces them with young cells with adequate G6PD.
G6PD Mediterranean variant (Middle East) produces more severe enzyme deficiency → more severe hemolysis.
Features in females: Heterozygous females may be symptomatic depending on pattern of X-inactivation ("unfavorable lyonization").
5. Paroxysmal Nocturnal Hemoglobinuria (PNH)
Type: Acquired, intravascular hemolysis
Pathogenesis:
- Acquired somatic mutation in PIGA gene (X-linked) in early hematopoietic progenitor
- PIGA is required for synthesis of phosphatidylinositol glycan (PIG) - membrane anchor for many proteins
- PIGA-deficient clone produces cells that lack "PIG-tailed" proteins, including several that limit complement activity (CD55 and CD59)
- Red cells derived from this clone are hypersensitive to lysis by C5b-C9 membrane attack complex
Nocturnal hemolysis: During sleep, CO₂ retention → ↓ blood pH → enhanced complement fixation → hemolysis of complement-sensitive red cells → hemoglobinuria most prominent in the morning
Clinical features:
- Most present less dramatically with chronic anemia and iron deficiency (chronic intravascular hemolysis → iron loss in urine)
- Thrombosis is the most feared complication - frequently in abdominal vessels (portal vein, hepatic veins → Budd-Chiari syndrome)
- Association with aplastic anemia (may precede or follow PNH)
- Leukocytes and platelets share the same PIGA deficiency but are less sensitive to complement than red cells
6. Immunohemolytic Anemia (Autoimmune Hemolytic Anemia)
Caused by antibodies binding to red cell membrane antigens - may arise spontaneously or be induced by drugs.
Diagnosis: Direct Coombs (antiglobulin) test - patient's red cells are incubated with anti-human IgG or anti-complement antibodies → agglutination = positive (confirms Ig and/or complement coating on patient's RBCs).
| Type | Antibody | Temperature | Causes |
|---|---|---|---|
| Warm antibody | IgG (rarely IgA) | Active at 37°C | Idiopathic (>60%), SLE, B-cell neoplasms, drugs (α-methyldopa, penicillin) |
| Cold antibody | IgM | Active at cold temps | Acute: Mycoplasma, EBV; Chronic: idiopathic, lymphoplasmacytic lymphoma |
Warm Antibody Immunohemolytic Anemia
- IgG autoantibodies bind red cells → removed by phagocytes in spleen (extravascular hemolysis)
- Macrophages also "nibble" membrane from antibody-coated cells → spherocyte formation → rapid splenic destruction
- Clinical: usually chronic mild anemia, moderate splenomegaly
Drug-induced mechanisms:
- Autoantibody induction (α-methyldopa): drug alters immunogenicity of native epitopes (e.g., Rh antigens) → bypasses T-cell tolerance
- Neoantigen (penicillin): drug binds covalently to red cell membrane proteins → antibodies form against drug-protein adduct
- Immune complex (quinidine): drug-antibody complexes deposit on red cells → complement fixation or opsonization → hemolysis
Cold Antibody Immunohemolytic Anemia
- IgM autoantibodies bind red cells at cold temperatures (e.g., extremities)
- As blood returns to warm core → IgM falls off but may have already fixed complement → complement-mediated lysis
7. Mechanical Hemolysis
Microangiopathic hemolytic anemia (MAHA):
- Red cells are physically fragmented as they flow through abnormal blood vessels or over artificial surfaces
- Schistocytes (helmet cells/fragmented cells) on peripheral smear are the hallmark
- Causes: TTP (thrombotic thrombocytopenic purpura), HUS (hemolytic uremic syndrome), DIC, prosthetic heart valves, severe hypertension, vasculitis
- The fragmented cells have reduced surface area-to-volume ratio → destroyed in spleen
8. Malaria
- Plasmodium parasites infect red cells → rupture of infected cells at end of each schizogony cycle
- Fever spikes correspond to synchronous rupture of parasitized erythrocytes
- Severe cases (esp. P. falciparum) → massive hemolysis → "blackwater fever" (hemoglobinuria)
- Splenomegaly from phagocytosis of infected red cells
Laboratory Features Summary
| Test | Extravascular | Intravascular |
|---|---|---|
| Serum bilirubin (indirect) | ↑↑ | ↑ (mild) |
| Haptoglobin | ↓ | ↓↓ |
| Hemoglobin in plasma | Normal | ↑ |
| Hemoglobin in urine | Absent | Present |
| Hemosiderin in urine | Absent | Present (chronic) |
| LDH | ↑ | ↑↑ |
| Reticulocytes | ↑ | ↑ |
| Splenomegaly | Common | Less prominent |
| Iron deficiency | Absent | Present (if chronic) |
Key Peripheral Blood Smear Findings
| Morphology | Associated Condition |
|---|---|
| Spherocytes (dark, no central pallor) | Hereditary spherocytosis, warm AIHA |
| Sickle cells (crescent-shaped) | Sickle cell anemia |
| Target cells (codocytes) | Thalassemia, HbC disease, liver disease |
| Bite cells + Heinz bodies | G6PD deficiency |
| Schistocytes (helmet cells) | Microangiopathic hemolytic anemia |
| Nucleated RBCs + teardrop cells | β-Thalassemia major |
| Basophilic stippling | Thalassemia, lead poisoning |
Sources: Robbins & Kumar Basic Pathology (Robbins Pathology), Chapter 10 - Hematopoietic and Lymphoid Systems, pp. 384-392
This is a shared conversation. Sign in to Orris to start your own chat.