Explain the mechanism of acute respiratory distress syndrome
Reading File
Finding Sources
Searching PubMed
"acute respiratory distress syndrome" AND pathogenesis
Reading File
Reading File
Reading File
Finding Sources
Reading File
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Finding Sources
Reading File
Loading Image
Loading Image
I now have comprehensive, authoritative content from Harrison's 22e, Murray & Nadel's, Robbins, and Sabiston. I have the mechanism diagram and the phase timeline. Let me now compose the full answer.
Acute Respiratory Distress Syndrome (ARDS): Mechanism
ARDS is a clinical syndrome of acute hypoxemic respiratory failure caused by diffuse alveolar damage (DAD) - a non-cardiogenic, inflammatory breakdown of the alveolar-capillary barrier. It is defined by the Berlin criteria: bilateral infiltrates, PaO2/FiO2 ratio <300, onset within 7 days of a known insult, and pulmonary edema not fully explained by heart failure or fluid overload.
Precipitating Triggers
ARDS arises from direct (pulmonary) or indirect (extrapulmonary) insults:
| Direct | Indirect |
|---|---|
| Pneumonia (bacterial, viral, fungal) | Sepsis (most common overall cause) |
| Aspiration of gastric contents | Severe trauma / polytrauma |
| Inhalation injury | Acute pancreatitis |
| Pulmonary contusion | Massive blood transfusion (TRALI) |
| Near-drowning | Hemorrhage / hypotension |
Pathophysiology: Three Sequential Phases
The time course of ARDS follows three overlapping stages:

Phase 1: Exudative Phase (Days 0-7)
This is the core injurious phase. The sequence of events:
1. Initial insult activates innate immunity
- Bacteria, viruses, or systemic mediators activate Toll-like receptors (TLRs) on alveolar type I (ATI) epithelial cells and resident alveolar macrophages
- Macrophages release proinflammatory cytokines: IL-1, IL-6, IL-8 (CXCL8), and TNF-α
- IL-8 is a potent neutrophil chemoattractant, driving massive pulmonary neutrophil recruitment
2. Neutrophil sequestration and activation
- Neutrophils (PMNs) migrate from pulmonary capillaries across the endothelium and epithelium into the alveolar space - this is the central step in injury
- Activated neutrophils release:
- Proteases (elastase, collagenase, MMP-8/9) - degrade basement membrane
- Reactive oxygen species (ROS) - cause oxidative injury to lipid membranes
- Neutrophil extracellular traps (NETs) - contribute to microvascular occlusion
- Platelet-activating factor - amplifies inflammatory signaling
3. Disruption of the alveolar-capillary barrier
- The barrier has two layers: the capillary endothelium and the alveolar epithelium (type I and II pneumocytes)
- Protease and ROS attack disrupts tight junctions and destroys type I pneumocytes (thin, gas-exchange cells covering ~95% of alveolar surface)
- Result: markedly increased permeability allows protein-rich exudative fluid to flood the interstitium and alveolar spaces (unlike cardiogenic edema which is low-protein hydrostatic)
The diagram below illustrates all of these cellular and molecular events occurring simultaneously in the injured alveolus:
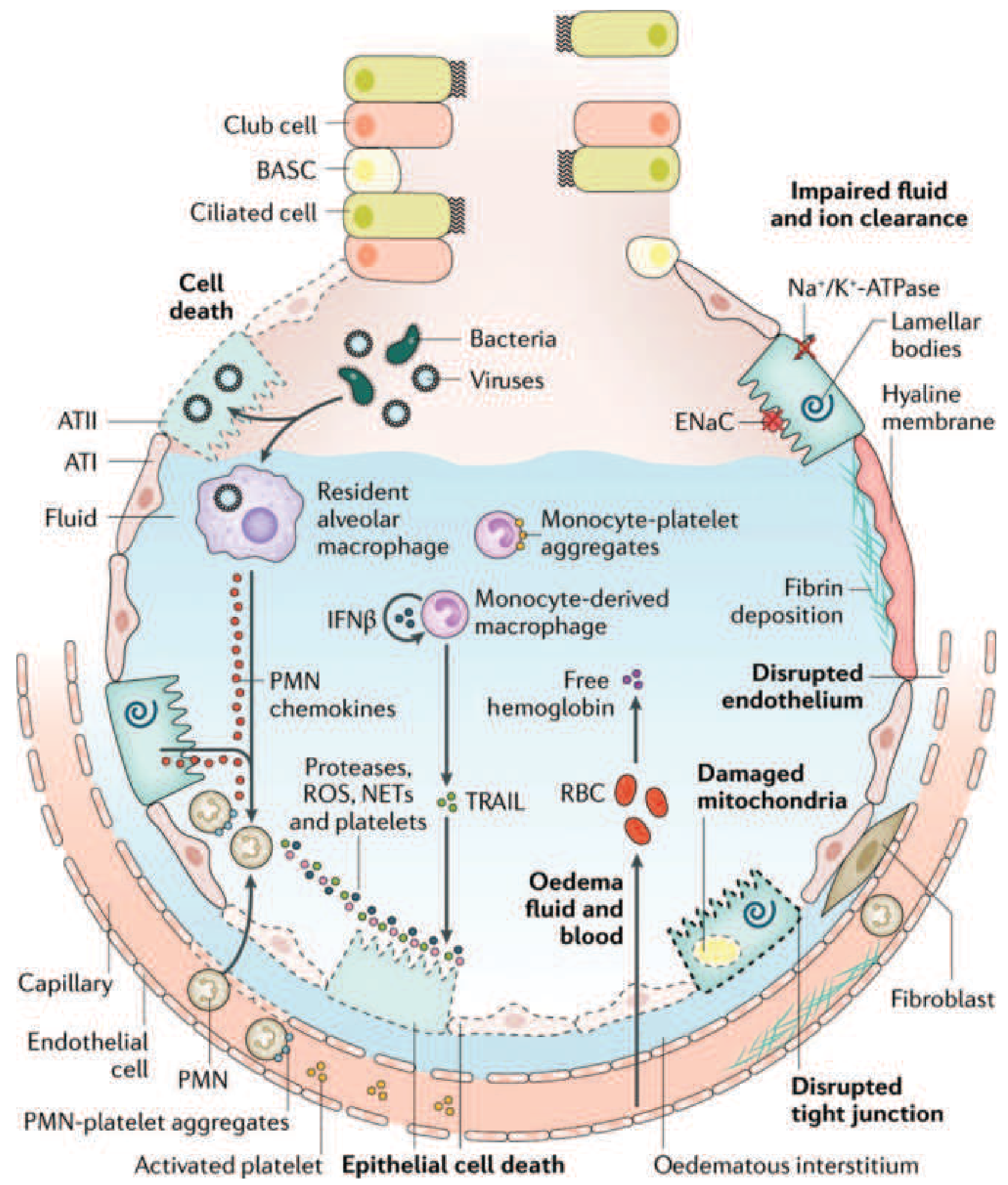
4. Monocyte-mediated epithelial apoptosis
- Circulating monocytes migrate into the lung and differentiate into macrophages
- They release IFN-β, which triggers monocyte-derived secretion of TRAIL (TNF-related apoptosis-inducing ligand)
- TRAIL activates death receptors on alveolar epithelial cells, causing apoptosis and further barrier breakdown
- PMN-platelet aggregates form and worsen NET formation
5. Red cell extravasation and oxidant injury
- Disrupted tight junctions allow RBCs into the alveolar space
- Lysed RBCs release cell-free hemoglobin, which generates additional oxidative stress via Fenton chemistry
6. Surfactant dysfunction
- Type II pneumocytes are also injured, reducing surfactant production
- Phospholipase A2 (elevated in pancreatitis-related ARDS) enzymatically degrades existing surfactant
- Loss of surfactant raises surface tension → alveolar collapse → atelectasis
7. Hyaline membrane formation
- Condensed plasma proteins, cellular debris, and dysfunctional surfactant components aggregate in the alveolar space to form characteristic hyaline membranes (named for their glassy histologic appearance)
8. Microvascular injury and pulmonary hypertension
- Vascular injury leads to microthrombosis (fueled by NETs and platelet aggregates) and fibrocellular proliferation in vessel walls
- Results in: dead-space ventilation, pulmonary vascular resistance, and pulmonary hypertension
- Mechanisms of pulmonary hypertension: hypoxic vasoconstriction + intravascular fibrin deposition + compression by positive-pressure ventilation
9. Impaired edema clearance
- Normally, alveolar fluid is cleared by ENaC (epithelial sodium channel) and Na+/K+-ATPase on type II pneumocytes
- Hypoxemia and hypercapnia directly impair sodium transport, so edema clearance is reduced, worsening the cycle
Net physiologic consequences:
- Alveolar edema (gravity-dependent, heterogeneous distribution) → ↓ functional residual capacity
- Collapsed dependent zones → intrapulmonary right-to-left shunt → refractory hypoxemia (hallmark)
- Dead space ventilation in non-collapsed regions → hypercapnia
- Reduced lung compliance (stiff lung) → increased work of breathing → respiratory fatigue and failure
Phase 2: Proliferative Phase (Days 7-21)
- The shift from neutrophil-predominant to lymphocyte-predominant inflammation signals the beginning of repair
- Type II pneumocytes (cuboidal, surfactant-producing) proliferate to cover denuded alveolar surfaces and differentiate into type I cells
- Hyaline membranes are reorganized and early fibrosis appears
- Fibroblast activation and collagen deposition begin (procollagen peptide III detectable in BAL fluid as early as 24 hours after onset - suggesting fibroproliferation begins simultaneously with injury)
- Many patients recover during this phase; some develop progressive lung injury
Phase 3: Fibrotic Phase (>Day 21)
- Occurs in a subset of patients with persistent ARDS (>2 weeks)
- Obliteration of pulmonary capillaries, extensive interstitial and alveolar collagen deposition, and bullae formation
- Loss of normal lung architecture
- Associated with prolonged mechanical ventilation dependence, increased risk of pneumothorax, and high mortality
Pathologic Hallmark: Diffuse Alveolar Damage (DAD)
DAD is the histologic correlate of ARDS - but importantly, only ~50% of ARDS patients show DAD on autopsy or biopsy. Patients with confirmed DAD tend to be younger, more severely ill, with lower compliance and P/F ratios, and are ~5x more likely to die of hypoxemic respiratory failure. The remaining 50% show other pathologies (pneumonia, diffuse alveolar hemorrhage, acute eosinophilic pneumonia, drug pneumonitis).
Histologic findings in DAD include:
- Alveolar edema
- Type I pneumocyte necrosis
- Neutrophil accumulation in interstitium and alveoli
- Hyaline membrane formation
- Interstitial and alveolar collagen (later stages)
Summary of Key Mediators
| Mediator | Source | Role |
|---|---|---|
| IL-1, IL-6 | Macrophages | Amplify systemic and local inflammation |
| IL-8 / CXCL8 | Macrophages, epithelium | Neutrophil chemoattraction |
| TNF-α | Macrophages | Endothelial activation, neutrophil priming |
| Leukotriene B4 | Macrophages, neutrophils | Neutrophil chemotaxis |
| Elastase, MMPs | Neutrophils | Barrier degradation |
| ROS | Neutrophils, macrophages | Oxidative membrane damage |
| NETs | Neutrophils | Microvascular occlusion, NETosis |
| TRAIL | Monocyte-derived macrophages | Epithelial apoptosis |
| Phospholipase A2 | Pancreas (pancreatitis) | Surfactant degradation |
| TGF-β | Multiple | Fibroblast activation, fibrosis |
What Makes It Non-Cardiogenic
Unlike left heart failure where elevated hydrostatic pressure drives low-protein fluid into alveoli, ARDS involves:
- Normal or low pulmonary capillary wedge pressure (<18 mmHg)
- High-protein exudative edema (reflecting barrier permeability, not hydrostatic pressure)
- Absence of cardiomegaly, pleural effusions, or pulmonary vascular redistribution on CXR (though these can coexist)
Sources:
- Harrison's Principles of Internal Medicine 22e, Chapter 312 - "Clinical course and pathophysiology"
- Murray & Nadel's Textbook of Respiratory Medicine - "Etiology and Pathogenesis of ARDS / Pathology / Alveolar-Capillary Membrane"
- Robbins & Kumar Basic Pathology - "Acute Lung Injury and Acute Respiratory Distress Syndrome"
- Sabiston Textbook of Surgery - "ARDS" (Berlin definition, management framework)
This is a shared conversation. Sign in to Orris to start your own chat.