Mi
It looks like your message got cut off! Could you finish your thought? What would you like help with?
Myocardial infarction
Asking for Preferences
Reading File
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Loading Image
Reading File
Finding Sources
Reading File
Reading File
Loading Image
Reading File
Loading Image
Reading File
I now have all the content I need to write a thorough overview and pathophysiology answer. Let me compile everything:
Myocardial Infarction - Overview & Pathophysiology
Definition
Myocardial infarction (MI) is necrosis of heart muscle resulting from ischemia. The 2018 Fourth Universal Definition (Joint ESC/ACC/AHA Task Force) defines it as "the presence of acute myocardial injury detected by abnormal cardiac biomarkers in the setting of evidence of acute myocardial ischemia."
- Robbins & Kumar Basic Pathology, p. 351
Epidemiology
In the United States, approximately 605,000 patients experience a new AMI and 200,000 experience a recurrent AMI each year. About half of AMI-related deaths occur before reaching the hospital. In-hospital mortality has declined from ~10% to ~5%. The 1-year post-AMI mortality rate is ~15%, and is approximately four-fold higher in patients aged >75 years.
- Harrison's Principles of Internal Medicine 22E, p. 2156
Pathogenesis: From Atherosclerosis to Occlusion
The progression from a normal vessel to an acute MI follows a well-defined sequence:
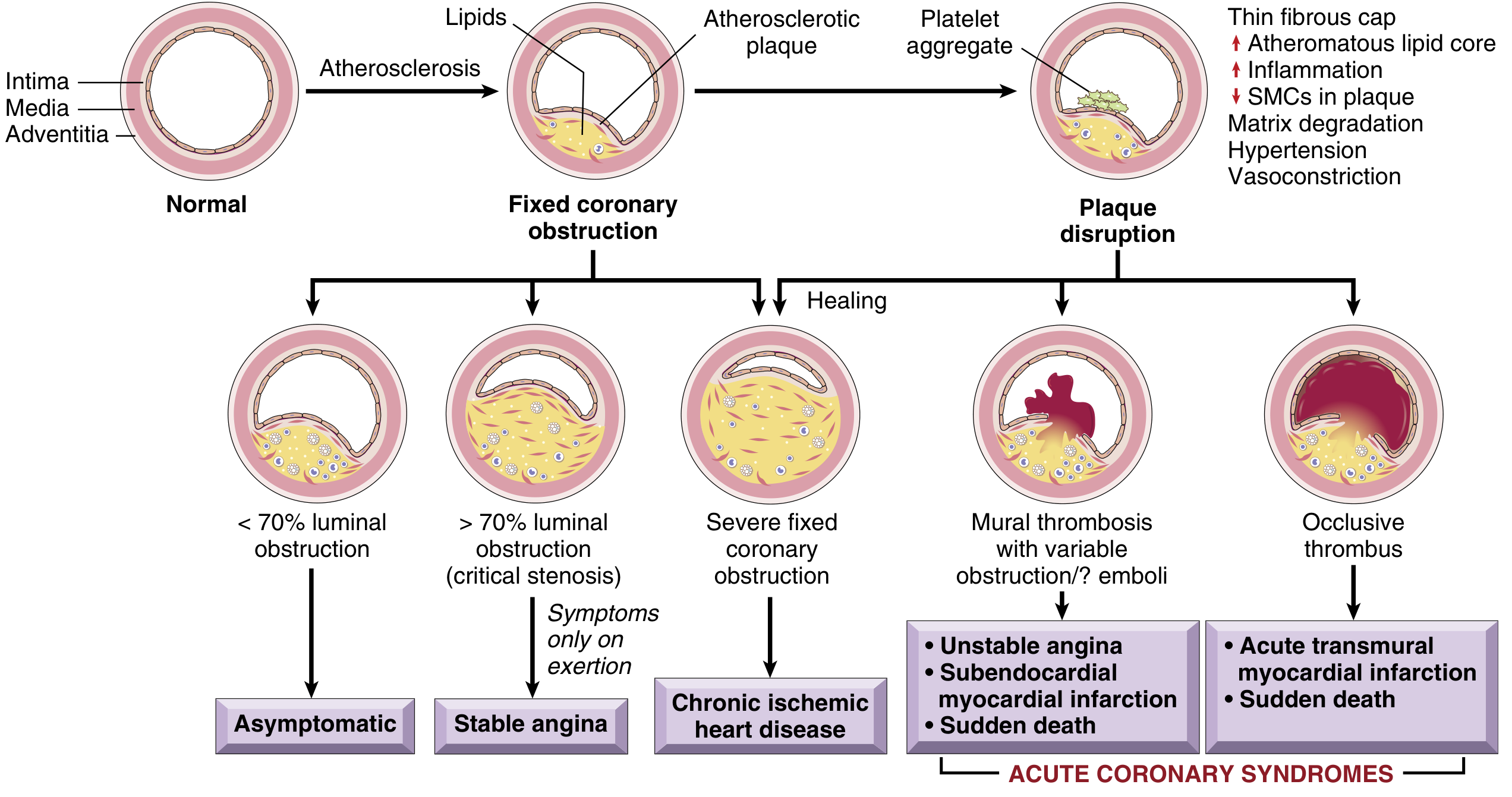
Fig. 9.7 - Sequential progression of coronary artery lesions. Plaque disruption with occlusive thrombus leads to acute transmural MI; mural thrombosis with partial obstruction leads to unstable angina/NSTEMI. (Robbins & Kumar Basic Pathology)
1. Atherosclerosis and Vulnerable Plaque
The vast majority of MIs arise from atherosclerotic coronary artery disease. Importantly, plaques prone to disruption have:
- A rich lipid core
- A thin fibrous cap (thin-cap fibroatheroma)
- Expansive remodeling, neovascularization (angiogenesis), and adventitial inflammation
- A "spotty" calcification pattern
Paradoxically, many MIs occur on plaques causing <70% luminal stenosis - these plaques were previously asymptomatic. Repetitive subclinical plaque ruptures with healing are also a mechanism by which plaques progressively enlarge.
2. Plaque Disruption
Two mechanisms of plaque disruption trigger acute MI:
- Rupture - mechanical cracking of the fibrous cap, exposing the lipid-rich, thrombogenic core to blood
- Erosion - superficial loss of endothelium without frank cap rupture (more common in younger women)
Risk factors that facilitate this disruption include cigarette smoking, hypertension, lipid accumulation, and inflammation.
3. Thrombosis
Once the plaque surface is disrupted:
- Platelet adhesion: A platelet monolayer forms at the site; agonists (collagen, ADP, epinephrine, serotonin) promote platelet activation
- Thromboxane A2 release: A potent local vasoconstrictor that promotes further platelet activation and resistance to fibrinolysis
- GP IIb/IIIa receptor activation: Converts to its functional state, binds fibrinogen (a multivalent molecule), causing platelet cross-linking and aggregation
- Coagulation cascade activation: Tissue factor exposure on damaged endothelium activates Factors VII and X → prothrombin → thrombin → fibrinogen → fibrin. Fluid-phase and clot-bound thrombin auto-amplify the cascade.
- Occlusive thrombus forms: containing platelet aggregates and fibrin strands - the final common pathway to STEMI
- Harrison's Principles of Internal Medicine 22E, pp. 2157-2158
4. Non-Atherosclerotic Causes (~10% of MIs)
- Vasospasm (Prinzmetal's) - with or without atherosclerosis; cocaine/ephedrine use
- Emboli - from mural left atrial thrombus (AF), infective endocarditis vegetations, or paradoxical emboli via PFO
- Spontaneous coronary artery dissection (SCAD)
- Small-vessel disease (vasculitis, amyloid, sickle cell)
- Global hypoperfusion (e.g., shock superimposed on 3-vessel disease)
Myocardial Response to Ischemia
Biochemical Cascade (Time-Dependent)
| Time After Onset | Event |
|---|---|
| Seconds | Cessation of aerobic metabolism; ATP and creatine phosphate depletion begins |
| ~1 minute | Loss of contractility (myocardium stops contracting before cells die) |
| Minutes | Ultrastructural changes: myofibrillar relaxation, glycogen depletion, cell and mitochondrial swelling; lactic acid accumulation |
| 20-30 min (severe ischemia <10% flow) | Irreversible injury (necrosis) begins |
| 3-6 hours | Progressive necrosis; full infarct extent reached |
| 6-12 hours | Nearly complete loss of viability in the affected zone |
The key functional consequence is that myocardial contractility ceases within a minute of severe ischemia - this is long before cell death occurs. This explains why large areas of "stunned" (reversibly injured) myocardium can recover if reperfusion is achieved promptly.
- Robbins, Cotran & Kumar Pathologic Basis of Disease, pp. 511-512
The Wavefront Phenomenon
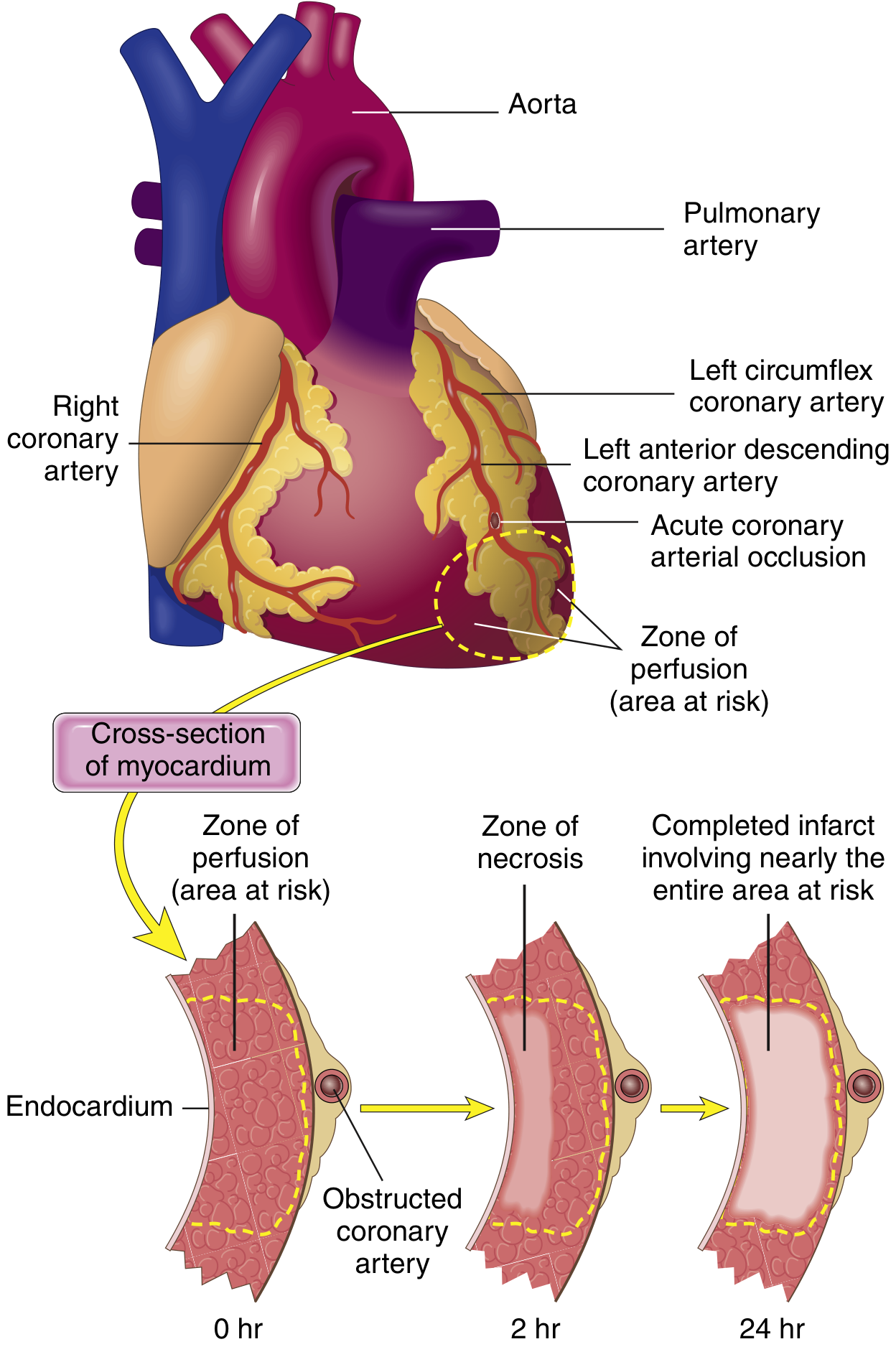
Fig. 9.8 - Necrosis begins in the subendocardial zone (most vulnerable) and expands as a wavefront over hours to involve the full wall thickness. A narrow rim beneath the endocardium is spared (oxygenated by diffusion from ventricular blood). (Robbins & Kumar Basic Pathology)
Irreversible ischemic injury first occurs in the subendocardium because:
- It is the last region to receive blood from epicardial vessels
- It is exposed to high intramural pressures that impede blood inflow
A wavefront of cell death then moves centripetally (outward, from endocardium toward epicardium), driven by progressive tissue edema, reactive oxygen species, and inflammatory mediators. Without intervention, full transmural necrosis is complete within 3-6 hours.
Clinical implication: This is the rationale for the time-critical nature of reperfusion therapy - every minute of delay extends the wavefront and permanently destroys more myocardium.
Infarction Patterns by Vessel Involved
| Vessel | Frequency | Territory Infarcted |
|---|---|---|
| LAD (proximal) | 40-50% | Anterior LV wall, anterior 2/3 of septum, LV apex |
| RCA (proximal) | 30-40% | Right ventricle, inferior/posterior LV |
| LCX (proximal) | 15-20% | Lateral LV wall |
Transmural vs. Subendocardial infarcts:
- Transmural: full-thickness necrosis from occlusive thrombus → associated with STEMI on ECG
- Subendocardial: limited to inner third, from partial/transient occlusion, global hypotension, or increased demand → associated with NSTEMI
Morphologic Changes Over Time
The infarcted zone progresses through a characteristic sequence:
| Time | Gross Appearance | Microscopic Changes |
|---|---|---|
| 0-12 hrs | Usually not grossly visible (TTC staining reveals unstained/pale areas) | Early coagulative necrosis begins; wavy fibers, cytoplasmic eosinophilia |
| 12-24 hrs | Red-blue discoloration (vascular engorgement) | Contraction band necrosis; PMN infiltration begins |
| 1-3 days | Yellow-tan (necrosis at center) | Dense PMN infiltration; myofiber loss |
| 3-7 days | Pallid/yellow, soft | Macrophage infiltration; phagocytosis of dead cells begins |
| 1-3 weeks | Red-gray periphery (granulation tissue) | Granulation tissue with new vessels and fibroblasts |
| Months | Gray-white, firm scar | Dense collagen scar - no regeneration occurs |
Myocardial necrosis proceeds invariably to scar formation with no significant myocyte regeneration, as cardiac myocytes are terminally differentiated cells.
- Robbins & Kumar Basic Pathology, p. 357
Biomarker Release
The earliest detectable feature of myocyte necrosis is disruption of the sarcolemmal membrane, allowing intracellular proteins to leak into the bloodstream. This forms the basis for:
- Troponin I and T (cTnI, cTnT) - rise within 3-4 hours, peak at 24-48 hours, remain elevated for 7-14 days
- CK-MB - rises within 4-6 hours, normalizes by 48-72 hours (useful for detecting reinfarction)
The temporal phase of the infarction must be considered when interpreting these tests.
- Harrison's Principles of Internal Medicine 22E, p. 2158
Electrical Consequences
Myocardial ischemia causes electrical instability in ischemic regions. While massive myocardial damage can cause fatal mechanical failure, 80-90% of cardiac deaths in the setting of ischemia are from ventricular fibrillation due to myocardial irritability - not pump failure. This is why defibrillation and arrhythmia management are so important in acute MI.
- Robbins & Kumar Basic Pathology, p. 355
Sources:
- Harrison's Principles of Internal Medicine 22E (2025), Chapter 286
- Robbins & Kumar Basic Pathology, Chapter 9
- Robbins, Cotran & Kumar Pathologic Basis of Disease, Chapter 12
This is a shared conversation. Sign in to Orris to start your own chat.