Provide me typesand subtypes of IBD and pathophysiology of IBD
Reading File
Finding Sources
Finding Sources
Reading File
Finding Sources
Finding Sources
Finding Sources
Reading File
Finding Sources
Finding Sources
Reading File
Finding Sources
Reading File
Reading File
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Reading File
Loading Image
I now have everything I need to deliver a comprehensive, well-illustrated answer. Let me compile the full response.
Inflammatory Bowel Disease (IBD): Types, Subtypes & Pathophysiology
1. Types and Subtypes of IBD
IBD is an umbrella term for chronic, relapsing-remitting inflammatory conditions of the GI tract. There are three major types:
A. Crohn's Disease (CD)
Crohn's disease can affect any segment of the GI tract from mouth to anus, with transmural inflammation and characteristic skip lesions.
Subtypes by anatomical location (Montreal Classification):
| Subtype | Location | Notes |
|---|---|---|
| Ileocolic (L3) | Terminal ileum + colon | Most common (~40%) |
| Ileal (L1) | Small intestine only | ~30% of cases |
| Colonic (L2) | Colon only | ~30%; hard to distinguish from UC |
| Upper GI (L4) | Esophagus, stomach, duodenum | <5%; usually with other disease |
| Perianal | Perianal region | Fistulas, abscesses, skin tags |
Subtypes by disease behavior (Vienna/Montreal Classification):
- B1 - Non-stricturing, non-penetrating (inflammatory): most common at diagnosis
- B2 - Stricturing: luminal narrowing from fibrosis, risk of obstruction
- B3 - Penetrating (fistulizing): deep fissures form fistulas (enteroenteric, enterovesical, perianal), abscesses, and perforation
Age at diagnosis modifier:
- A1: <16 years
- A2: 17-40 years
- A3: >40 years
B. Ulcerative Colitis (UC)
UC is confined strictly to the colon, with mucosal (non-transmural) continuous inflammation always beginning at the rectum and extending proximally.
Subtypes by extent (Montreal Classification):
| Subtype | Extent | Notes |
|---|---|---|
| E1 - Ulcerative Proctitis | Rectum only (≤15 cm) | Mildest; risk of CRC only slightly increased vs. general population |
| E2 - Left-sided colitis (Distal UC) | Up to splenic flexure | CRC risk begins ~1 decade later than pancolitis |
| E3 - Extensive colitis / Pancolitis | Beyond splenic flexure, entire colon | Highest CRC risk (34% at 30 years in some series); risk of toxic megacolon |
Subtypes by disease severity:
- Mild: <4 stools/day, minimal systemic signs, no anemia
- Moderate: 4-6 stools/day, mild systemic signs
- Severe (Truelove-Witts): >6 bloody stools/day with systemic toxicity (fever, tachycardia, anemia, elevated ESR/CRP)
- Fulminant: >10 bloody stools/day, continuous bleeding, abdominal tenderness - risk of toxic megacolon
C. IBD-Unclassified (IBDU) / Indeterminate Colitis
- Used when colonoscopic or histological findings do not clearly distinguish CD from UC, particularly in colonic disease
- Accounts for ~5-10% of IBD cases
- May be reclassified after further evaluation or resection specimen review
Comparison: CD vs. UC at a Glance
(Source: Goldman-Cecil Medicine, Robbins Pathologic Basis of Disease)
| Feature | Crohn's Disease | Ulcerative Colitis |
|---|---|---|
| Site | Esophagus to anus | Colon only |
| Distribution | Skip lesions | Continuous, from rectum proximally |
| Inflammation | Transmural | Mucosal only |
| Ulcers | Deep, knife-like / serpentine | Superficial, broad-based |
| Wall | Thickened, rubbery | Normal thickness |
| Granulomas | Yes (~35%) | No |
| Fistulae/Sinuses | Yes | No |
| Strictures | Common | Rare |
| Perianal disease | Yes | No |
| Fibrosis | Marked | Mild to none |
| Toxic megacolon | No | Yes |
| Recurrence after surgery | Common | No (colectomy is curative) |
| Cancer risk | With colonic involvement | Yes (pancolitis) |
2. Pathophysiology of IBD
IBD results from a combination of abnormalities in immune regulation, host-microbe interactions, and epithelial barrier function in genetically susceptible individuals - Robbins, Cotran & Kumar Pathologic Basis of Disease.
The central model is breakdown of homeostasis between three components:
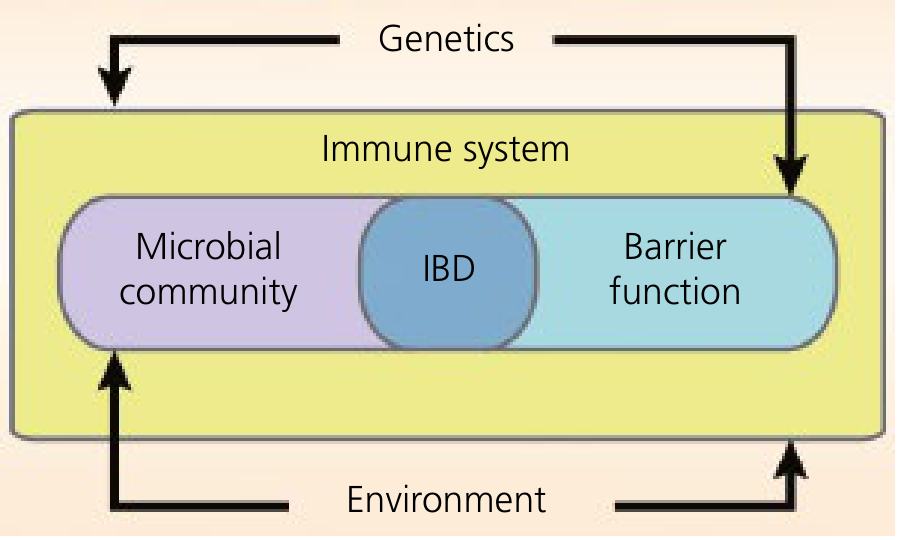
IBD arises at the intersection of dysbiosis (altered microbial community), impaired barrier function, and dysregulated immune system - all shaped by genetics and environment. (Yamada's Textbook of Gastroenterology)
A. Genetic Susceptibility
- Over 200 IBD-associated risk alleles have been identified; together they account for <20% of disease risk in CD and even less in UC - confirming IBD is a polygenic disorder
- NOD2 (chromosome 16): the most strongly associated gene with Crohn's disease. NOD2 encodes an intracellular sensor of muramyl dipeptide (MDP), a bacterial cell wall peptidoglycan. Three common polymorphisms (Arg702Trp, Gly908Arg, Leu1007fsX1008) are present in ~30% of CD patients in European populations but only ~5% of healthy individuals. NOD2 activation triggers NF-kB and MAPK signaling, producing TNF-α and IL-1β. Loss-of-function mutations impair bacterial clearance.
- ATG16L1: involved in autophagosome formation. The T300A mutation leads to abnormal Paneth cell granule formation and defective secretion of antimicrobial defensins.
- PTPN22: increases CD risk but is protective in UC - one of few genes differentially associated with the two diseases
- Genes at IL10/IL10RA/IL10RB loci: loss-of-function mutations here cause monogenic IBD (very-early-onset infantile colitis)
- IBD risk alleles overlap significantly with type 1 diabetes, ankylosing spondylitis, and psoriasis - reflecting shared immune mechanisms
B. Intestinal Epithelial Barrier Dysfunction
The gut epithelium maintains homeostasis through:
- Intercellular tight junctions and adherens junctions
- Mucus layer produced by goblet cells
- Antimicrobial peptides secreted by Paneth cells (defensins, lysozyme)
- Immune regulation by intestinal epithelial cells (IEC)
How it breaks down in IBD:
- IBD risk genes CDH1, HNF4A, GNA12, MUC19, ITLN1 all affect barrier integrity. For example, CDH1 polymorphisms produce a truncated E-cadherin → defective goblet cell and Paneth cell maturation → reduced clearance of pathogenic bacteria.
- PTPN2 and GNA12 maintain tight junction integrity; PTPN2-deficient mice develop colitis.
- NOD2 and ATG16L1 mutations → Paneth cell defects → reduced defensin secretion → impaired bacterial clearance
- Endoplasmic reticulum (ER) stress: XBP1 deletion causes spontaneous colitis; disruption of the unfolded protein response (UPR) increases susceptibility. This is particularly relevant to the secretory burden on Paneth cells and goblet cells.
- Even healthy first-degree relatives of CD patients show increased intestinal permeability - indicating barrier defects may be a primary predisposing factor, not just a consequence of inflammation.
C. Gut Microbiome and Dysbiosis
- The gut microbiome (bacteria, fungi, viruses) normally exists in homeostasis with the host immune system
- Patients with IBD consistently show dysbiosis - shifts in microbial populations and reduced species diversity
- Altered microbial metabolites (short-chain fatty acids, bile acids, tryptophan metabolites) affect immune and epithelial function
- The "hygiene hypothesis": improved sanitation and food storage have altered microbial exposures during early life, impairing development of regulatory immune processes - this may explain the rising IBD incidence in historically low-prevalence regions (Africa, South America, Asia)
- Dietary emulsifiers (polysorbate-80, carboxymethylcellulose) found in processed foods may disrupt the mucous barrier, contributing to dysbiosis and intestinal inflammation
D. Innate Immune Dysregulation
This is particularly central to Crohn's disease pathogenesis.
Key cells and mechanisms:
- Dendritic cells and macrophages sense microbiota via pattern recognition receptors (PRRs) recognizing pathogen-associated molecular patterns (PAMPs)
- Bacterial antigens are presented via HLA class II molecules on dendritic cells, activating the innate immune response
- NOD2 variants, ATG16L1, CARD9, IL18RAP, and SLC11A1 polymorphisms all lead to a defective innate immune response to luminal antigens
- Defective bacterial killing by neutrophils and macrophages in CD paradoxically prolongs the inflammatory stimulus - the gut cannot clear the inciting organisms
E. Adaptive Immune Dysregulation (T-Cell Polarization)
This is the central inflammatory mechanism driving tissue destruction.
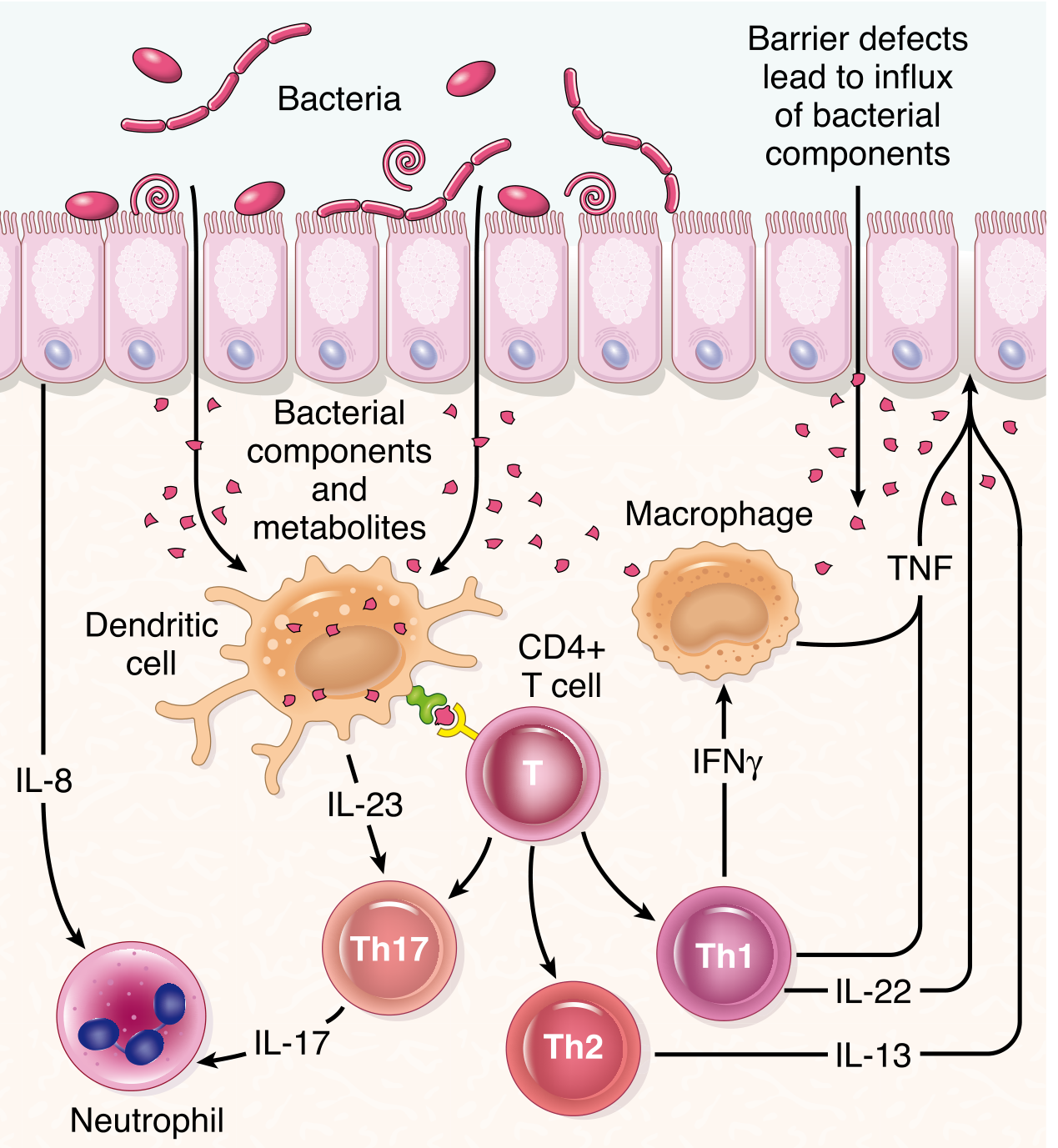
Barrier defects allow bacterial components to penetrate the epithelium, activating dendritic cells that drive CD4+ T-cell polarization into Th1, Th17, and Th2 subsets. Each secretes distinct proinflammatory cytokines, amplifying mucosal damage. (Robbins, Cotran & Kumar Pathologic Basis of Disease)
In Crohn's disease:
- Dominant Th1 response: dendritic cell-derived IL-12 → Th1 polarization → IFN-γ and TNF-α production → macrophage activation and granuloma formation
- Th17 response: dendritic cell-derived IL-23 → Th17 polarization → IL-17, IL-22, IL-21 → neutrophil recruitment and epithelial damage
In Ulcerative Colitis:
- More prominent Th2 component: → IL-13 and IL-5 → epithelial barrier damage (IL-13 disrupts tight junctions)
- Th1/Th17 responses also occur
Regulatory T-cell (Treg) failure:
- In the steady state, tolerogenic DCs (CD103+) produce IL-10 and IL-27, expanding Tregs that suppress Th1/Th17
- In IBD, this regulatory circuit fails: Tregs are numerically reduced or functionally impaired → Th1/Th17 dominate → chronic inflammation perpetuates
Key cytokines and their roles:
| Cytokine | Source | Effect |
|---|---|---|
| TNF-α | Macrophages, Th1 | Master proinflammatory mediator; drives NF-kB, tissue damage |
| IL-12 | Dendritic cells | Drives Th1 polarization |
| IL-23 | Dendritic cells | Drives Th17 polarization |
| IFN-γ | Th1 cells | Activates macrophages, barrier disruption |
| IL-17 | Th17 cells | Neutrophil recruitment |
| IL-13 | Th2 cells | Barrier dysfunction (UC) |
| IL-10 | Tregs, macrophages | Anti-inflammatory; loss → colitis |
F. Autophagy Defects
- ATG16L1 and IRGM variants impair autophagosome formation
- Autophagy normally eliminates damaged organelles, clears reactive oxygen species, and kills intracellular pathogens
- In CD, the ATG16L1 Crohn-associated mutation accelerates protein degradation → impaired autophagy → structurally and functionally abnormal Paneth cell granules → reduced antimicrobial defensin secretion → persistent intracellular bacteria → sustained inflammation
Summary: The Unified Pathophysiological Model
- Genetic susceptibility (NOD2, ATG16L1, HLA, PTPN22, IL10, etc.) creates baseline vulnerability
- Environmental triggers (diet, hygiene, antibiotics, smoking, stress) alter the gut microbiome → dysbiosis
- Barrier dysfunction (leaky tight junctions, defective mucus, impaired Paneth cells) allows luminal antigens to translocate into the mucosa
- Antigen exposure activates innate immune cells (dendritic cells, macrophages) via NOD2 and other PRRs
- Ineffective bacterial clearance (defective autophagy) sustains the antigenic stimulus
- Innate cells prime CD4+ T cells → predominantly Th1/Th17 polarization (with some Th2 in UC)
- Proinflammatory cytokines (TNF-α, IFN-γ, IL-17, IL-12/23) drive transmural (CD) or mucosal (UC) inflammation
- Regulatory T-cell failure prevents resolution → chronic relapsing inflammation
- Chronic inflammation causes crypt destruction, fibrosis (CD), mucosal ulceration, and ultimately complications (strictures, fistulas in CD; toxic megacolon, dysplasia in UC)
Key sources:
- Robbins, Cotran & Kumar Pathologic Basis of Disease - Pathogenesis section, Ch. 17
- Yamada's Textbook of Gastroenterology, 7th ed. - Ch. 62 (Intestinal epithelial barrier, immune response)
- Goldman-Cecil Medicine International Edition - Ch. 127 (Clinical characteristics table, UC/CD morphology)
- Sleisenger and Fordtran's Gastrointestinal and Liver Disease - Ch. 115 (CD activity, disease patterns)
This is a shared conversation. Sign in to Orris to start your own chat.