what is first pass metabolism in pharmacology
Reading File
Finding Sources
Searching the Web
first pass metabolism pharmacology mechanism bioavailability
Finding Sources
Reading File
Reading File
Reading File
Loading Image
Loading Image
Here is a thorough breakdown of first pass metabolism, drawn from Lippincott Pharmacology, Katzung, and Goodman & Gilman:
First Pass Metabolism (First Pass Effect)
Definition
First pass metabolism (also called the first pass effect or presystemic metabolism) is the biotransformation of a drug before it reaches systemic circulation. When a drug is swallowed and absorbed from the GI tract, it enters the portal vein and travels directly to the liver. The liver - and sometimes the gut wall itself - can metabolize a significant fraction of the drug during this initial transit, so that less active drug actually reaches the bloodstream and the target tissues.
"If the drug is rapidly metabolized in the liver or gut wall during this initial passage, the amount of unchanged drug entering the systemic circulation is decreased." - Lippincott Illustrated Reviews: Pharmacology
How It Happens - Step by Step
- Drug is swallowed and absorbed through the intestinal wall into mesenteric capillaries
- These drain into the portal vein, which goes directly to the liver
- Hepatic enzymes (primarily CYP450 enzymes, especially CYP3A4) metabolize a portion of the drug
- The gut wall also contributes (e.g., CYP3A4 and P-glycoprotein efflux)
- Only the fraction that survives this first passage enters the hepatic vein → inferior vena cava → systemic circulation
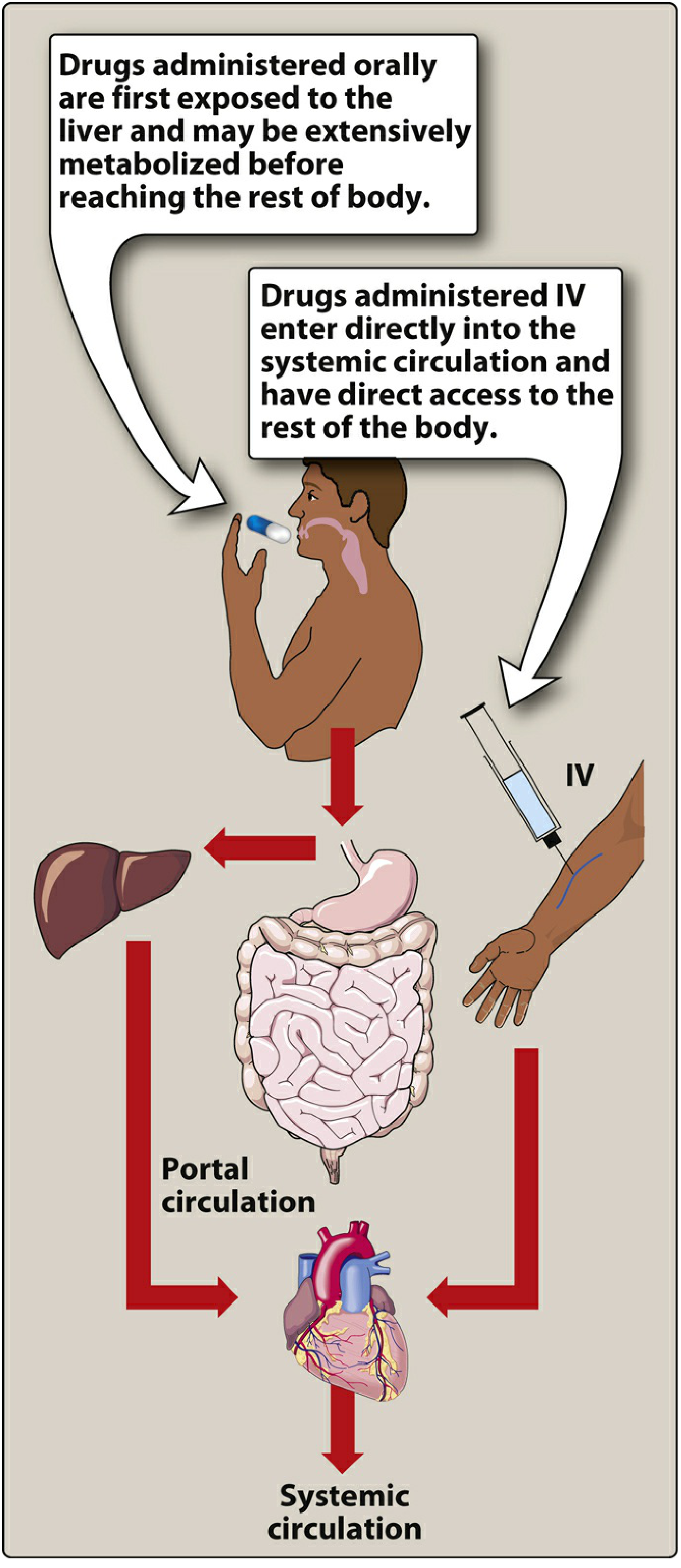
Figure: Oral drugs are exposed to the liver via portal circulation before reaching the body. IV drugs bypass this entirely. (Lippincott Pharmacology)
Effect on Bioavailability
Bioavailability (F) is the fraction of an administered dose that reaches systemic circulation unchanged. First pass metabolism is one of the key reasons oral bioavailability can be much lower than IV bioavailability.
F = AUC(oral) / AUC(IV) × 100%
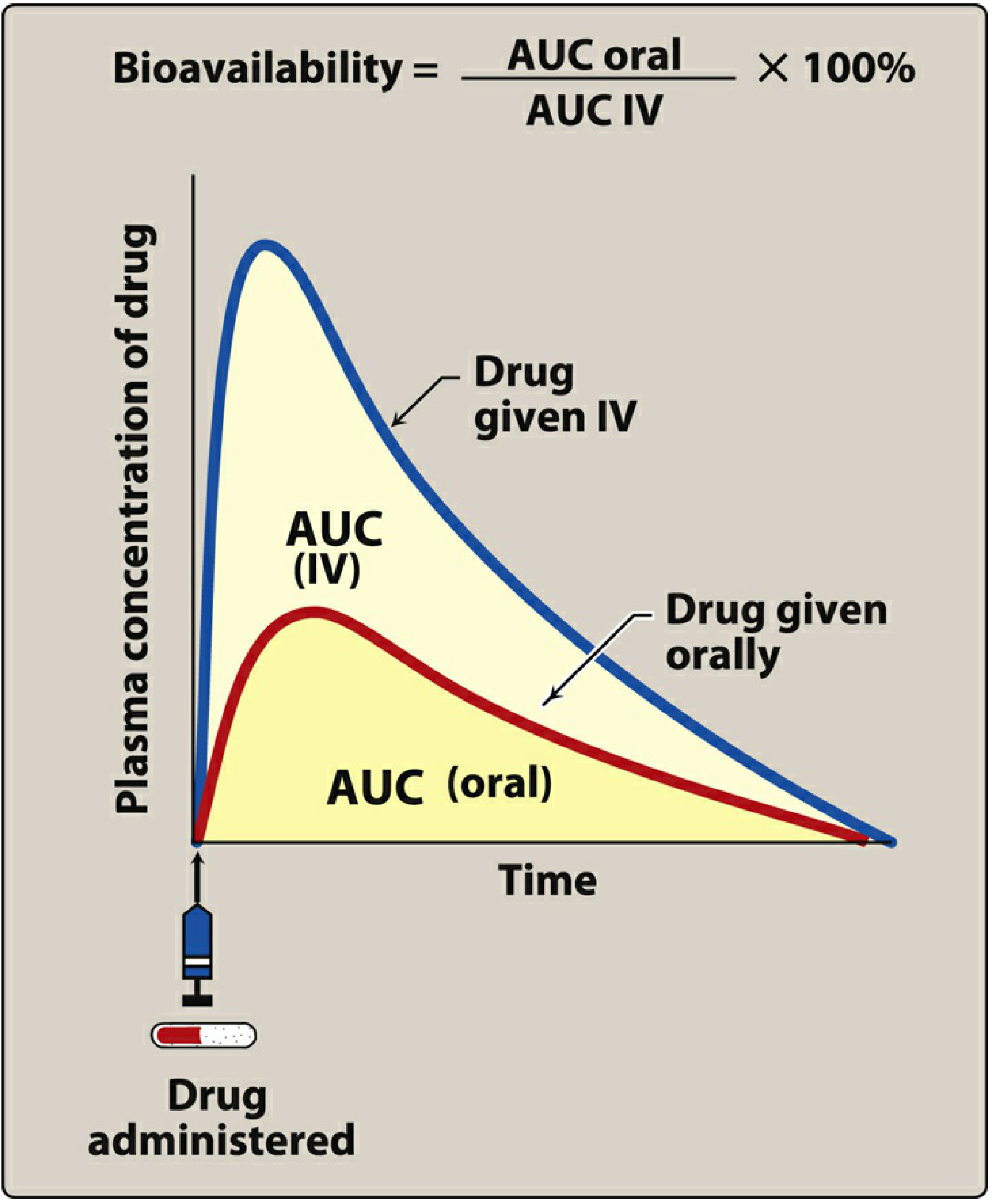
Figure: Oral drug (red curve) achieves a lower AUC than IV (blue curve) due to first pass metabolism. (Lippincott Pharmacology)
The extraction ratio is the proportion of drug removed by the liver per pass. Drugs with a high extraction ratio have very low oral bioavailability.
Classic Examples
| Drug | First Pass Effect | Clinical Implication |
|---|---|---|
| Nitroglycerin | >90% cleared | Must be given sublingually, transdermally, or IV - NOT orally |
| Lidocaine | Extensive (>60%) | Given IV only; oral metabolites cause CNS toxicity |
| Morphine | Significant | Oral dose must be much higher than IV dose |
| Propranolol | High | Large oral doses needed; wide interpatient variability |
| Naloxone | Extensive | Ineffective orally; given IV or IM |
| Isoniazid, Tricyclic antidepressants | High | Dose adjustments required |
| Codeine, Oxycodone | Low | Effective orally precisely because of reduced first pass |
Sites of First Pass Metabolism
The liver is the primary site, but first pass can occur in multiple locations:
- Gut wall - CYP3A4 and P-glycoprotein (e.g., benzylpenicillin, insulin degraded here)
- Liver - Main site; CYP450 enzymes (propranolol, lidocaine, nitroglycerin, morphine)
- Lung - For drugs given by non-GI parenteral routes; can serve as a site of first pass loss via excretion or metabolism
Routes That Bypass First Pass Metabolism
| Route | Bypass Mechanism |
|---|---|
| Intravenous (IV) | Enters systemic circulation directly |
| Sublingual | Absorbed into systemic veins (not portal), e.g., nitroglycerin |
| Transdermal | Bypasses portal circulation entirely |
| Inhalation | Bypasses hepatic first pass (though lungs may metabolize some drugs) |
| Rectal (suppository) | Lower rectum drains into inferior vena cava (~50% bypass); upper rectum drains into portal circulation |
Clinical Relevance
- Dose adjustment: Drugs with extensive first pass need much higher oral doses than IV doses (e.g., morphine oral:IV ratio is approximately 3:1)
- Inter-patient variability: Differences in CYP450 activity, hepatic blood flow, and genetic polymorphisms cause wide variation between individuals
- Liver disease: Cirrhosis with portosystemic shunting can dramatically increase bioavailability of high first-pass drugs (e.g., morphine, meperidine, midazolam, nifedipine oral bioavailability nearly doubles in cirrhosis - per Harrison's Principles of Internal Medicine)
- Drug interactions: One drug can inhibit or induce the enzymes involved, affecting the first pass metabolism of another drug (e.g., grapefruit juice inhibits CYP3A4)
- Prodrugs: Some drugs are designed to exploit first pass - they are inactive until metabolized by the liver (e.g., codeine → morphine via CYP2D6; enalapril → enalaprilat)
Summary Formula
Oral bioavailability (F) = Fraction absorbed × (1 - Extraction Ratio)
A drug with 100% GI absorption but a 0.9 extraction ratio ends up with only 10% bioavailability.
Sources: Lippincott Illustrated Reviews: Pharmacology | Katzung's Basic and Clinical Pharmacology, 16th Ed | Goodman & Gilman's The Pharmacological Basis of Therapeutics | Goldman-Cecil Medicine | Harrison's Principles of Internal Medicine 22E
Add a NOTE on DRUG ABSORPTION & FACTORS influencing it?
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Finding Sources
Reading File
Loading Image
Now I have everything needed for a thorough, well-illustrated note. Here it is:
NOTE: Drug Absorption & Factors Influencing It
What is Drug Absorption?
Absorption is the transfer of a drug from its site of administration into the systemic circulation. It is the first step in the pharmacokinetic sequence - ADME (Absorption, Distribution, Metabolism, Excretion).
"Absorption from the site of administration permits entry of the drug (either directly or indirectly) into plasma." - Lippincott Illustrated Reviews: Pharmacology
Most drugs cross biological membranes by passive diffusion - moving down a concentration gradient. The rate depends on the concentration of free (unbound, uncharged) drug at the absorbing surface.
Mechanisms of Drug Absorption Across Membranes
| Mechanism | Energy | Saturable? | Notes |
|---|---|---|---|
| Passive diffusion | No | No | Most common; uncharged, lipophilic drugs prefer this |
| Facilitated diffusion | No | Yes | Uses carrier proteins; large molecules |
| Active transport | Yes (ATP) | Yes | Against concentration gradient; competitive inhibition possible |
| Endocytosis | Yes | No | Very large molecules (e.g., Vitamin B12 across gut wall) |
Factors Influencing Drug Absorption
1. pH and Drug Ionization (pKa)
This is one of the most important factors. Most drugs are weak acids or weak bases and exist in both ionized (charged) and un-ionized (uncharged) forms depending on the pH of the surrounding environment.
- Un-ionized form = lipid soluble → crosses membranes freely
- Ionized form = water soluble, charged → does NOT cross lipid membranes well
Henderson-Hasselbalch principle:
- Weak acids (e.g., aspirin, warfarin): better absorbed in acidic environments (stomach) where they are un-ionized
- Weak bases (e.g., morphine, codeine, atropine): better absorbed in alkaline environments (small intestine) where they are un-ionized
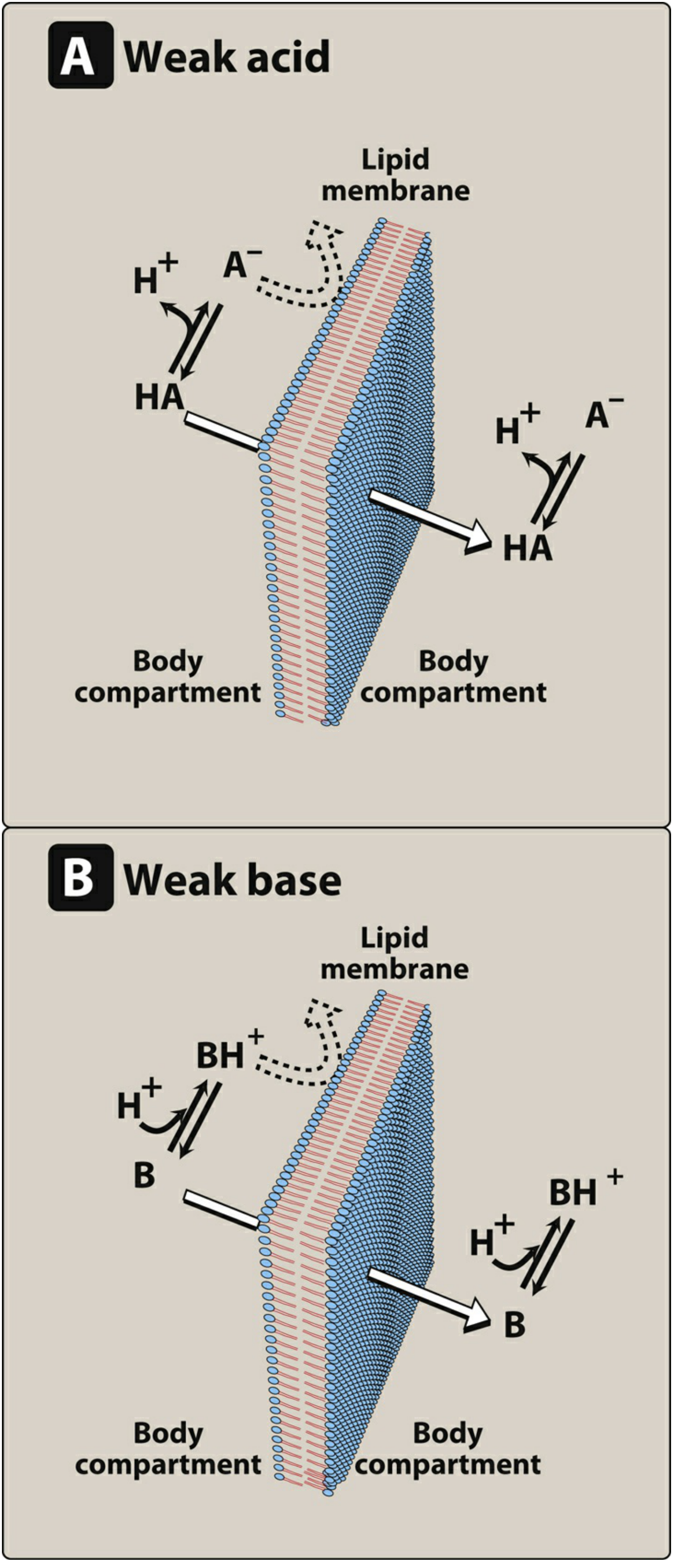
Figure: Only the uncharged (non-ionized) form of a drug permeates the lipid membrane. For weak acids (HA), the neutral form crosses; for weak bases (B), the free base form crosses. (Lippincott Pharmacology)
Ion trapping: When a drug passes into a compartment where it becomes ionized, it gets "trapped" there - it cannot diffuse back out. This is exploited clinically, e.g., alkalinizing urine to trap acidic drugs like aspirin for faster excretion in overdose.
2. Lipid Solubility
- Drugs must be lipophilic enough to dissolve in the lipid bilayer and cross membranes
- But must also have some water solubility to dissolve in GI fluids and reach the membrane surface
- Extremely hydrophilic OR extremely lipophilic drugs are both poorly absorbed
- This is why many drugs are weak acids or weak bases - they have partial lipid solubility
3. Route of Administration
The route determines which absorption barriers a drug faces and whether it undergoes first pass metabolism.
| Route | Onset | First Pass? | Notes |
|---|---|---|---|
| IV | Immediate | No | 100% bioavailability; gold standard |
| Sublingual/Buccal | Fast (minutes) | No | Directly into systemic veins |
| Oral | Slow-moderate | Yes | Most complex; affected by GI factors |
| Transdermal | Slow | No | Bypasses portal circulation |
| Inhalation | Fast | Minimal | Large surface area, rich blood supply |
| Rectal | Variable | ~50% bypass | Unpredictable absorption |
| IM / SC | Moderate | No | Depot effect possible |
4. GI Motility and Gastric Emptying
- Increased GI motility (e.g., diarrhea, metoclopramide) → faster gastric emptying → drug reaches small intestine sooner → may increase absorption rate but reduce total time for absorption
- Decreased motility (e.g., opioids, anticholinergics) → delayed gastric emptying → slower absorption onset
- Most oral absorption occurs in the small intestine (large surface area, rich vascular supply), so faster gastric emptying generally increases drug absorption
5. Blood Flow to the Absorption Site
- Greater blood flow maintains a high concentration gradient across the membrane, driving faster absorption
- IM injections absorb faster than SC because muscle has richer blood supply
- In shock states, peripheral absorption (IM, SC) is unreliable due to vasoconstriction - IV is preferred
- Sustained-release and depot preparations slow drug release to prolong absorption regardless of blood flow
6. Solubility and Drug Formulation
"Drug absorption may be altered by factors unrelated to the chemistry of the drug. For example, particle size, salt form, crystal polymorphism, enteric coatings, and the presence of excipients (such as binders and dispersing agents) can influence the ease of dissolution and absorption." - Lippincott Pharmacology
Key formulation factors:
- Particle size: smaller particles dissolve faster (more surface area)
- Salt form: e.g., sodium salts dissolve faster than free acid forms
- Crystal polymorphism: different crystal forms of the same drug dissolve at different rates
- Enteric coatings: protect acid-labile drugs (e.g., omeprazole) or irritating drugs (e.g., aspirin EC) - release occurs in the intestine
- Extended-release (ER/SR/XL) formulations: rate of dissolution controls rate of absorption; allows twice or once-daily dosing
7. Food and Drug Interactions
- Food can delay gastric emptying, dilute drug concentration, or chelate certain drugs (e.g., tetracyclines bind calcium in dairy)
- P-glycoprotein (P-gp): an efflux transporter in the gut wall that pumps some drugs back into the intestinal lumen, reducing absorption (e.g., digoxin). Inhibitors like verapamil increase digoxin bioavailability
- Grapefruit juice: inhibits intestinal CYP3A4 → increases absorption of many drugs (e.g., statins, calcium channel blockers)
- Antacids: raise gastric pH, altering ionization of drugs (e.g., reduces absorption of ketoconazole which needs acidic environment)
8. Drug Chemical Instability
- Some drugs are degraded before absorption:
- Penicillin G: unstable in acidic gastric environment
- Insulin: proteolytic enzymes in the GI tract destroy it completely - must be given by injection
- Nitroglycerin: destroyed by hepatic first pass (>90%) if swallowed
9. Patient-Specific Factors
| Factor | Effect on Absorption |
|---|---|
| Liver disease (cirrhosis) | Reduced first pass → higher bioavailability of high-extraction drugs |
| Age (elderly/neonates) | Altered gastric pH, GI motility, blood flow |
| CKD | Reduced first pass metabolism of some beta-blockers |
| Bowel disease (Crohn's, celiac) | Mucosal damage reduces absorption surface area |
| Obesity (post-bariatric surgery) | Malabsorptive procedures (e.g., RYGB) markedly alter drug absorption |
| Gender | Differences in GI motility and CYP enzyme activity affect bioavailability |
Summary Diagram of Key Concepts
Drug administered orally
↓
Dissolved in GI fluids (solubility)
↓
Un-ionized form (depends on pKa + pH) crosses gut wall
↓
P-gp efflux? CYP3A4 in gut wall? → reduced absorption
↓
Portal vein → LIVER → first pass metabolism
↓
Remaining drug → systemic circulation = BIOAVAILABILITY (F)
Sources: Lippincott Illustrated Reviews: Pharmacology | Goldman-Cecil Medicine | Katzung's Basic and Clinical Pharmacology, 16th Ed | Harrison's Principles of Internal Medicine 22E
This is a shared conversation. Sign in to Orris to start your own chat.