Plase tell me about hypokalemia from harrsion 22 refrence and fialis textbook of nephrology
Reading File
Finding Sources
Finding Sources
Finding Sources
Reading File
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
I now have all the information needed to provide a comprehensive answer. Let me compile it:
Hypokalemia
Harrison's Principles of Internal Medicine, 22nd Edition (2025)
Definition & Epidemiology
Hypokalemia is defined as a plasma K+ concentration of <3.5 mM (mEq/L). It occurs in up to 20% of hospitalized patients and is associated with a tenfold increase in in-hospital mortality, due to adverse effects on cardiac rhythm, blood pressure, and cardiovascular morbidity.
- Harrison's Principles of Internal Medicine, 22E, Ch. 56
Causes of Hypokalemia (Harrison's Table 56-4)
Hypokalemia can result from three broad mechanisms:
1. Decreased Intake
- Starvation
2. Redistribution into Cells (Transcellular Shift)
Acid-base:
- Metabolic alkalosis
Hormonal:
- Insulin (especially exogenous insulin - iatrogenic)
- Increased β2-adrenergic sympathetic activity: post-MI, head injury
- β2-adrenergic agonists - bronchodilators, tocolytics (ritodrine)
- α-adrenergic antagonists
- Thyrotoxic periodic paralysis (TPP)
- Downstream stimulation of Na+/K+-ATPase: theophylline, caffeine overdose
Anabolic states:
- Vitamin B12 or folic acid administration (RBC production)
- GM-CSF (WBC production)
- Total parenteral nutrition
Other:
- Pseudohypokalemia
- Hypothermia
- Familial hypokalemic periodic paralysis
- Barium toxicity (inhibits leak K+ channels)
3. Increased Loss
Nonrenal:
- GI loss (diarrhea)
- Integumentary loss (sweat)
Renal:
- Increased distal flow: diuretics, osmotic diuresis, salt-wasting nephropathies
- Mineralocorticoid excess: primary hyperaldosteronism (adenoma, hyperplasia, carcinoma), secondary hyperaldosteronism (malignant HTN, renin-secreting tumors, renal artery stenosis), Cushing's syndrome, Bartter's syndrome, Gitelman's syndrome
- Apparent mineralocorticoid excess (AME), Liddle syndrome
- Non-reabsorbable anions: bicarbonaturia, hippurate, penicillinase
- Hypomagnesemia
- Tubular toxins: amphotericin B, cisplatin, aminoglycosides
Redistribution Mechanisms (Harrison's, detailed)
Insulin, β2-adrenergic activity, thyroid hormone, and alkalosis all promote Na+/K+-ATPase-mediated cellular uptake of K+. Exogenous insulin is a frequent cause of iatrogenic hypokalemia (notably in DKA management). β2-agonists are powerful activators of cellular K+ uptake.
Thyrotoxic Periodic Paralysis (TPP):
- Attacks of profound hypokalemia with weakness, most frequent between 1-6 AM
- Hypokalemia is usually profound and accompanied by hypophosphatemia and hypomagnesemia
- More common in patients of Asian or Latin American origin
- Linked to genetic variation in Kir2.6 (muscle-specific, thyroid hormone-responsive K+ channel)
- Pathomechanism: direct and indirect activation of Na+/K+-ATPase + reduced outward KIR current
- Treatment: high-dose propranolol (3 mg/kg) rapidly reverses hypokalemia WITHOUT risk of rebound hyperkalemia (unlike aggressive K+ replacement, which carries ~25% incidence of rebound hyperkalemia)
Familial Hypokalemic Periodic Paralysis: Missense mutations in voltage sensor domains within the α subunit of L-type calcium channels or skeletal Na+ channel.
Nonrenal Potassium Loss
- Sweat: Typically minimal
- Vomiting/NGT suctioning: Direct gastric K+ loss is minimal; the ensuing hypochloremic alkalosis causes persistent kaliuresis via secondary hyperaldosteronism and bicarbonaturia
- Diarrhea: Quantitatively the most important nonrenal cause; associated with non-anion gap metabolic acidosis
Diagnostic Approach (Harrison's Figure 56-7)
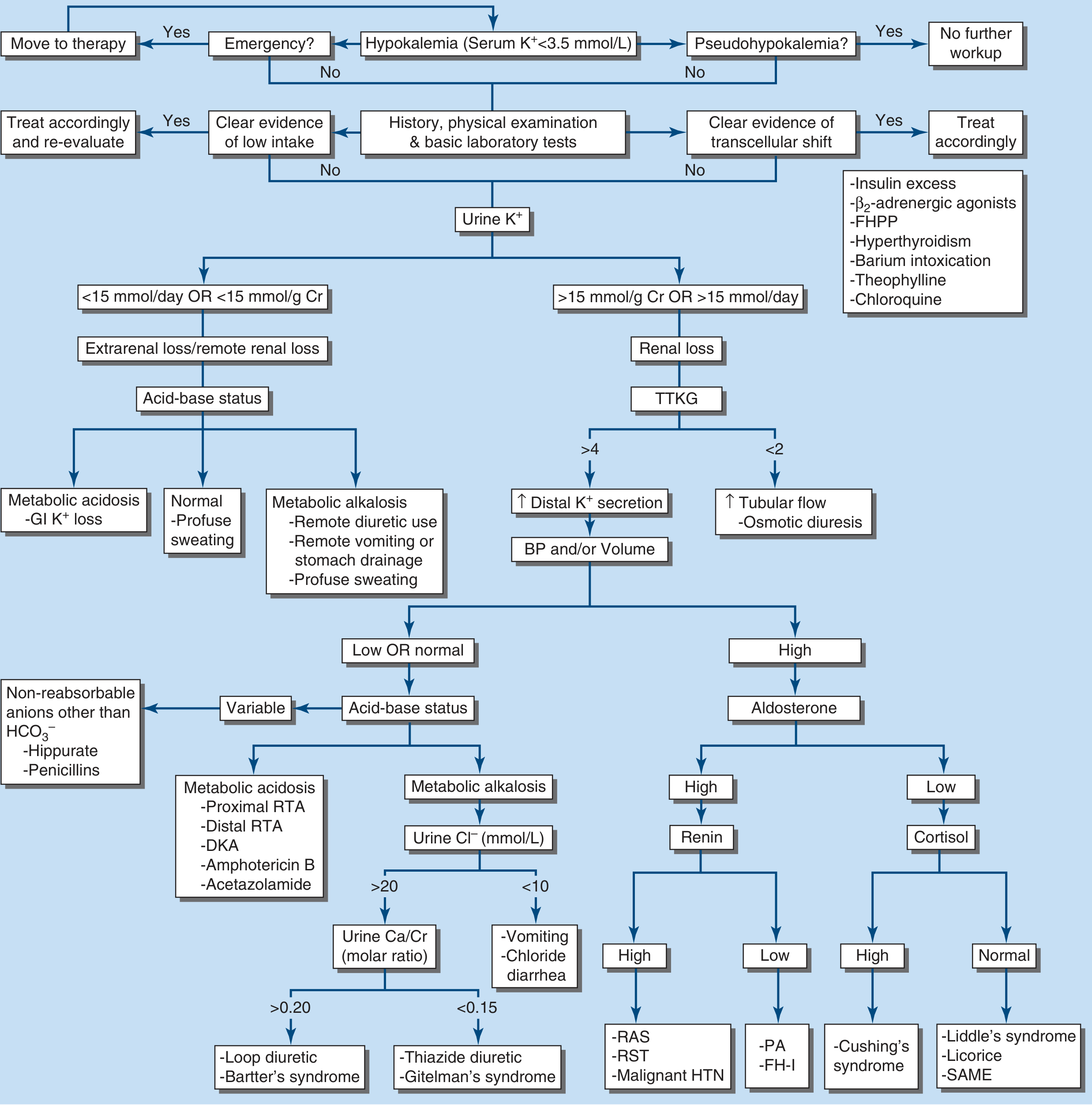
Key steps:
- Is it a true emergency? → Move to therapy directly
- Rule out pseudohypokalemia
- History, physical, and basic labs: look for low intake or transcellular shift
- Check urine K+:
- <15 mmol/day or <15 mmol/g Cr → Extrarenal loss
- >15 mmol/day or >15 mmol/g Cr → Renal loss
- For renal loss: assess TTKG
- TTKG >4 → Increased distal K+ secretion → assess BP and volume → if high BP/aldosterone:
- High renin: RAS, renin-secreting tumor, malignant HTN
- Low renin, high aldosterone: Primary aldosteronism, FH-1
- Low renin, low aldosterone, high cortisol: Cushing's
- Low renin, low aldosterone, normal cortisol: Liddle's, licorice, SAME
- TTKG <2 → Increased tubular flow, osmotic diuresis
- TTKG >4 → Increased distal K+ secretion → assess BP and volume → if high BP/aldosterone:
- For extrarenal loss: check acid-base:
- Metabolic acidosis: GI K+ loss (diarrhea)
- Metabolic alkalosis: remote diuretic use, vomiting, NG drainage
- Normal: profuse sweating
Treatment (Harrison's)
Goals: Prevent life-threatening complications, replace K+ deficit, correct the underlying cause.
Urgency depends on:
- Severity of hypokalemia
- Cardiac disease, digoxin therapy
- Rate of decline
Key principles:
| Situation | Action |
|---|---|
| Prolonged QT or arrhythmia risk | Continuous cardiac telemetry during repletion |
| Severe redistributive hypokalemia (<2.5 mM) with serious complications | Urgent but cautious K+ replacement |
| TPP or sympathomimetic-driven redistribution | Propranolol 3 mg/kg (avoids rebound hyperkalemia) |
| Hypomagnesemia | Must correct Mg2+ first - patients are refractory to K+ replacement alone |
Oral K+ replacement:
- KCl is the mainstay
- K+ phosphate if combined hypokalemia + hypophosphatemia
- K+ bicarbonate or citrate if combined metabolic acidosis
Estimating the deficit:
- Serum K+ drops by ~0.27 mM per 100-mmol reduction in total body stores
- Loss of 400-800 mmol of total body K+ reduces serum K+ by ~2.0 mM
- Replace gradually over 24-48 hours with frequent monitoring (delay in redistribution into intracellular compartment)
Intravenous K+:
- Reserve for patients unable to use enteral route, or severe complications (paralysis, arrhythmia)
- IV KCl should be given via peripheral vein at rates not exceeding 10-20 mEq/h to avoid cardiac toxicity
- Concentrations >40 mEq/L require central venous access
Brenner & Rector's The Kidney (Fialis Textbook of Nephrology equivalent - Comprehensive Clinical Nephrology)
Epidemiology (Brenner)
- ~20% of hospitalized patients have serum K+ <3.6 mmol/L
- 16.8% of first-time hospital admissions have K+ <3.4 mmol/L
- Usually mild (K+ 3.0-3.5 mmol/L), but up to 25% of hypokalemic patients have moderate-severe levels (≤3.0 mmol/L)
- Most common causes in hospitalized patients: GI losses, diuretic therapy, hypomagnesemia
- Thiazide diuretics: incidence 15-30% (up to 48%)
- Metolazone: moderate hypokalemia (K+ ≤3.0) in ~40%, severe (K+ ≤2.5) in ~10%
- Can increase in-hospital mortality up to tenfold
Spurious (Pseudohypokalemia)
- Delayed sample analysis at high ambient temperature → increased cellular uptake of K+
- Profound leukocytosis (acute leukemia): K+ is taken up by the large white cell mass in vitro
- Key diagnostic clue: no clinical or ECG changes of hypokalemia; K+ normal if measured immediately after venipuncture
- Management: rapid plasma separation and storage at 4°C
Redistribution Causes (Brenner)
- Administered insulin: frequent cause of iatrogenic hypokalemia; may contribute to "dead in bed syndrome" with aggressive glycemic control; also important in refeeding syndrome
- Sympathetic nervous system activation: alcohol withdrawal, acute MI, head injury (can be profound - reported K+ of 1.2-1.9 mmol/L after severe head injury)
- Thyrotoxic Periodic Paralysis (TPP): K+ ranges 1.1-3.4 mmol/L; TTKG <2-3 or urine K+:creatinine <2.5 mmol/mmol distinguishes from renal wasting (TTKG >4); aggressive KCl replacement carries ~25% risk of rebound hyperkalemia in TPP
Diuretic-Induced Hypokalemia (Brenner)
Four mechanisms increase renal K+ elimination with thiazides or loop diuretics:
- Increased tubular flow - flow-dependent K+ secretion via BK (big K+) channels
- AVP secretion - nonosmotic AVP release (common in edema) + increased distal flow → ongoing K+ losses
- Aldosterone secretion - diuretic-induced RAAS activation
- Alkalosis - bicarbonaturia enhances K+ secretion
Basal K+ secretion: mediated by ROMK channels
Flow-dependent K+ secretion: mediated by BK (calcium-activated maxi K+) channels
Prevention/treatment strategies: direct renin inhibitors, ACEi/ARBs, mineralocorticoid receptor antagonists (spironolactone), ENaC blockers (amiloride, triamterene), KCl supplements.
Hypokalemia in Cancer Patients (Brenner)
Unique causes:
- Tubular damage from chemotherapy (cisplatin, ifosfamide) or antimicrobials (amphotericin B, aminoglycosides)
- Light-chain-induced proximal tubulopathy (multiple myeloma)
- Ectopic ACTH syndrome - excessive cortisol overwhelms 11β-HSD-2, creating apparent mineralocorticoid excess → HTN + severe hypokalemia
- Lysozymuria (hematologic malignancies)
- Chemotherapy-induced GI losses
Ectopic ACTH: Most common causes are bronchial carcinoid tumors, small cell lung carcinoma, thymic cancers, pancreatic tumors. K+ in ectopic ACTH syndrome is more severely depressed than in Cushing's disease (pituitary ACTH) - incidence 57-100% vs. 10%.
Syndromes of Apparent Mineralocorticoid Excess (AME) - Brenner
Classic AME: recessive loss-of-function mutations in the 11β-HSD-2 gene → defective conversion of cortisol to cortisone → cortisol acts on mineralocorticoid receptor (which has equal affinity for cortisol and aldosterone) → hypertension, hypokalemia, metabolic alkalosis with suppressed PRA and aldosterone.
Diagnosis: urinary free cortisol to free cortisone ratio on 24-hour urine collection.
Similar mechanism in licorice ingestion (glycyrrhizic acid inhibits 11β-HSD-2) and Liddle syndrome (gain-of-function ENaC mutations).
Hypokalemia in Pediatric Patients (Brenner, Ch. 73)
Causes:
| Acquired | Inherited |
|---|---|
| Metabolic alkalosis, insulin, hypokalemic periodic paralysis | Bartter syndrome, Gitelman syndrome, EAST syndrome |
| Diuretics, antibiotics (aminoglycosides, amphotericin B, capreomycin) | Fanconi syndrome, RTA types 1/2/3 |
| Vomiting, HPS, diarrhea | CAH, primary hyperaldosteronism, Liddle syndrome, AME |
Clinical manifestations:
- Neuromuscular: weakness, paralysis, paralytic ileus, rhabdomyolysis
- Cardiac: ST depression, low T wave amplitude, U wave appearance, arrhythmias
- Nutrition/Growth: mild glucose intolerance, growth retardation
- Kidney: hypokalemic nephropathy, polyuria-polydipsia
Summary Comparison Table
| Feature | Harrison's 22E | Brenner & Rector's Kidney |
|---|---|---|
| Definition | <3.5 mM | <3.5-3.6 mmol/L |
| Prevalence in hospitalized | Up to 20% | ~20% (up to 48% on thiazides) |
| Mortality impact | 10-fold increase | 10-fold increase |
| TPP treatment | Propranolol 3 mg/kg | Propranolol; distinguish TTKG <2-3 (redistribution) vs >4 (renal wasting) |
| K+ deficit formula | ↓0.27 mM per 100 mmol deficit | - |
| IV rate | ≤10-20 mEq/h | - |
| Diuretic mechanism | 4 mechanisms highlighted | Detailed ROMK vs BK channel distinction |
| Special populations | Detailed on TPP, familial periodic paralysis | Cancer, pediatrics, AME |
| Mg2+ correction | Mandatory before K+ repletion | Highlighted as most common cause with GI/diuretics |
References:
- Harrison's Principles of Internal Medicine, 22nd Edition (2025) - Chapter 56: Fluid and Electrolyte Disturbances (pp. 398-472)
- Brenner and Rector's The Kidney, 2-Volume Set - Chapters 10, 25, 30, 41 (Hypokalemia sections)
This is a shared conversation. Sign in to Orris to start your own chat.