Sideroblastic anemia
Reading File
Finding Sources
Searching PubMed
"sideroblastic anemia"[MeSH Terms]
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
I now have comprehensive information from multiple authoritative textbooks. Here is the full answer:
Sideroblastic Anemia
Definition
Sideroblastic anemias are a group of disorders defined by the presence of ringed sideroblasts in the bone marrow - erythroid precursors in which iron-laden mitochondria form a perinuclear ring, visible on Prussian blue stain. The underlying defect is disrupted heme synthesis, causing iron to accumulate in mitochondria rather than being incorporated into hemoglobin.
A ringed sideroblast is defined as an erythroblast with 5 or more iron-staining granules encircling at least one-third of the nuclear circumference.
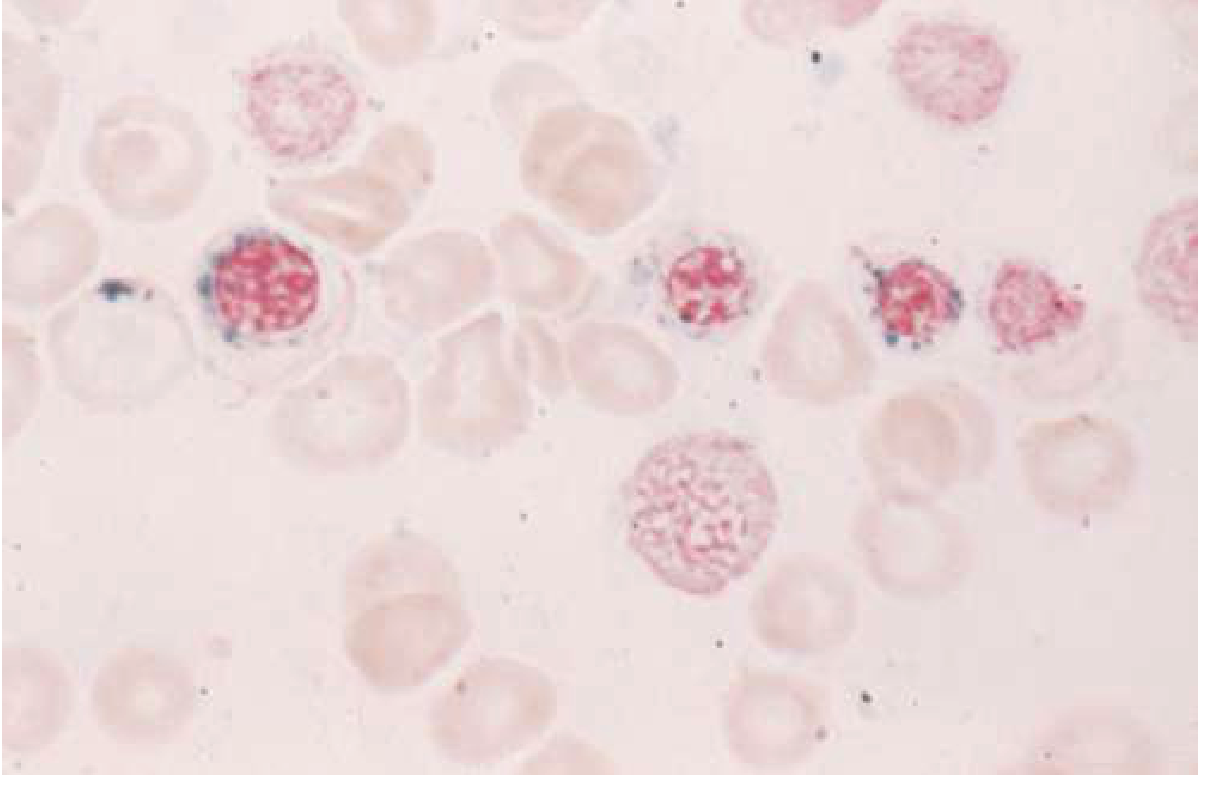
Prussian blue stain showing ringed sideroblasts (erythroblasts with perinuclear rings of iron-laden mitochondria). Source: Goldman-Cecil Medicine
Classification
1. Inherited (Congenital) Forms
| Type | Gene/Defect | Inheritance | Notes |
|---|---|---|---|
| X-linked sideroblastic anemia (XLSA) | ALAS2 mutation (erythroid delta-aminolevulinic acid synthase) | X-linked recessive | ~40% of hereditary cases; enzyme has low affinity for pyridoxal phosphate |
| SLC25A38 deficiency | Mitochondrial glycine transporter | Autosomal recessive | Impairs glycine import for heme synthesis |
| Glutaredoxin 5 deficiency | GLRX5 | Autosomal recessive | Impairs ALAS2 translation |
| Mitochondrial cytopathies | Mitochondrial genome deletions | Mitochondrial | Up to 30% of hereditary forms; systemic manifestations |
| Pearson syndrome | Mitochondrial DNA deletion | Mitochondrial | Sideroblastic anemia + exocrine pancreatic insufficiency + vacuolated bone marrow cells; usually macrocytic; can mimic Diamond-Blackfan anemia |
XLSA clinical features (Harrison's 22E):
- Males present in infancy with refractory hemolytic anemia, pallor, weakness
- Hypochromic, microcytic anemia with marked anisocytosis and poikilocytosis
- Secondary hypersplenism and iron overload/hemosiderosis
-
120 loss-of-function ALAS2 mutations identified
- Bimodal RBC volume distribution on peripheral blood
2. Acquired Forms
A. Myelodysplastic Syndrome with Ring Sideroblasts (MDS-RS)
- Most common acquired cause overall
- The majority harbor SF3B1 pathogenic mutations (a splicing factor gene)
- Usually normocytic to macrocytic anemia
B. Drug- and Toxin-Induced (reversible if agent removed)
- Alcohol - most common reversible cause; decreases pyridoxal phosphate, inhibits ALA dehydratase and ferrochelatase; ring sideroblasts disappear within days of abstinence
- Isoniazid (INH), pyrazinamide, cycloserine - anti-TB drugs that interfere with heme synthesis; respond to pyridoxine supplementation
- Linezolid - antibiotic
- Chloramphenicol - inhibits mitochondrial protein synthesis
- Lead poisoning - blocks ALAS, ALA dehydratase, and heme synthase; urinary ALA and coproporphyrin elevated
- Copper deficiency (dietary or from zinc overload/penicillamine) - causes ring sideroblasts, vacuolated marrow cells, and neutropenia
- Chemotherapy, irradiation
Pathophysiology
The core defect is failure of heme synthesis, with several possible enzyme blocks:
- ALAS2 deficiency (XLSA) - first and rate-limiting step in heme synthesis
- Glycine import failure (SLC25A38 defect)
- Enzyme inhibition by drugs (isoniazid inhibits PLP-dependent steps; lead blocks multiple enzymes)
- Mitochondrial protein synthesis inhibition (chloramphenicol, alcohol)
Iron that cannot be incorporated into heme accumulates in mitochondria, forming the perinuclear ring. Ineffective erythropoiesis occurs with intramedullary destruction of red cell precursors.
Laboratory Findings
Peripheral Blood
- Inherited forms: microcytic, hypochromic RBCs; bimodal (dimorphic) RBC size distribution
- Acquired forms: may be microcytic, normocytic, or macrocytic; a population of microcytic cells usually visible on smear
- Basophilic stippling and Pappenheimer bodies
- Anisocytosis, poikilocytosis
Bone Marrow
- Ringed sideroblasts (Prussian blue stain)
- Erythroid hyperplasia with dyserythropoiesis
- Increased iron stores
- (Occasionally megaloblast-like changes, but without granulocyte changes of B12/folate deficiency)
Serum Studies
- Serum iron: elevated
- TIBC: decreased
- Transferrin saturation: greatly elevated (mimics hereditary hemochromatosis)
- Ferritin: elevated
- Bilirubin: elevated (indirect) - from intramedullary hemolysis
- LDH: elevated
- Haptoglobin: low
Diagnosis
Definitive diagnosis requires bone marrow examination showing ringed sideroblasts. Once drugs and toxins are excluded, molecular diagnostics can identify the specific inherited mutation (e.g., ALAS2, SF3B1). In copper deficiency, neurologic manifestations (myelopathy) are often present.
Treatment
| Category | Treatment |
|---|---|
| XLSA (pyridoxine-responsive) | High-dose pyridoxine (vitamin B6) - works when mutation disrupts pyridoxal phosphate binding; may eliminate or reduce transfusion need |
| XLSA (pyridoxine-unresponsive) | Chronic transfusions + chelation therapy (to manage iron overload) |
| Drug/toxin-induced | Remove the offending agent; alcohol-induced resolves rapidly with abstinence and nutrition |
| MDS-RS | Treat underlying MDS (luspatercept, ESAs, transfusions depending on risk) |
| Inherited (severe) | Allogeneic stem cell transplantation in eligible patients |
| Iron overload (all forms) | Chelation therapy (deferoxamine, deferasirox) |
Key Pearls
- Dimorphic blood smear (mixed micro/normocytic population) is a classic finding - especially in inherited forms and after partial treatment
- All sideroblastic anemias share elevated serum iron and high transferrin saturation despite anemia (distinguish from iron deficiency where these are low)
- SF3B1 mutation is the molecular hallmark of MDS-RS (acquired form)
- Pearson syndrome = sideroblastic anemia + pancreatic exocrine insufficiency (mitochondrial deletion)
- Pyridoxine responsiveness depends on whether the ALAS2 mutation disrupts the pyridoxal phosphate binding site
- Copper deficiency (from zinc excess - e.g., excess zinc supplement use or denture adhesive ingestion) is an underrecognized reversible cause
Sources: Goldman-Cecil Medicine (27th ed.), Henry's Clinical Diagnosis and Management by Laboratory Methods, Harrison's Principles of Internal Medicine (22nd ed., 2025), Quick Compendium of Clinical Pathology (5th ed.)
This is a shared conversation. Sign in to Orris to start your own chat.