Chronic myeloid leukemia
Reading File
Finding Sources
Searching PubMed
"chronic myeloid leukemia" AND treatment
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Reading File
Reading File
Loading Image
Loading Image
Reading File
Excellent - I now have comprehensive content covering pathogenesis, morphology, clinical features, phases, TKI therapy, milestones, and prognosis. I have enough to write a thorough, well-structured response.
Chronic Myeloid Leukemia (CML)
Overview
Chronic myeloid leukemia is a clonal hematopoietic stem cell neoplasm and the prototypical myeloproliferative disorder. Its entire pathobiology is driven by a single defining molecular event: the BCR::ABL1 fusion oncogene. Before tyrosine kinase inhibitors (TKIs), the median survival was 3-7 years; today, with modern TKI therapy, the 10-year survival exceeds 85% and approaches that of an age-matched general population.
Molecular Pathogenesis
The defining event is a reciprocal translocation between chromosomes 9 and 22: t(9;22)(q34.1;q11.2), creating the Philadelphia (Ph) chromosome (shortened chromosome 22). This juxtaposes the ABL1 proto-oncogene from chromosome 9 with the BCR gene on chromosome 22, generating the chimeric BCR::ABL1 oncogene.
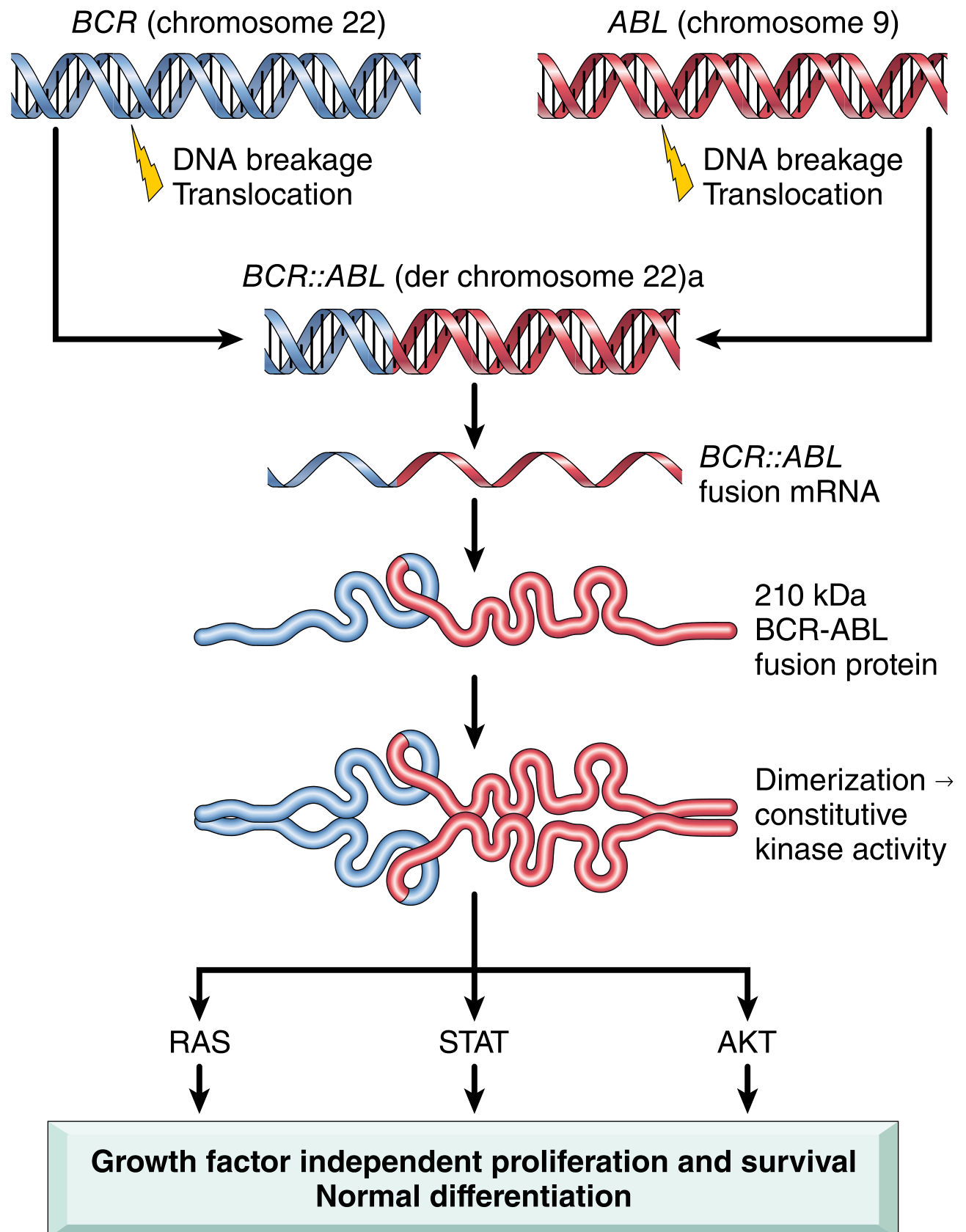
The BCR moiety contains a dimerization domain that self-associates, leading to constitutive activation of the ABL tyrosine kinase. This oncoprotein (p210 BCR-ABL, 210 kDa) then phosphorylates downstream targets activating the RAS, JAK/STAT, and AKT pathways - driving growth factor-independent proliferation and survival while preserving normal differentiation. The net result is a massive increase in mature granulocytes and platelets in the blood. The cell of origin is a pluripotent hematopoietic stem cell (HSC), explaining why both myeloid and lymphoid lineages can be involved.
- The Ph chromosome is present in >90% of cases; the remaining ~10% have cryptic BCR::ABL1 rearrangements detectable only by FISH or PCR.
- Two main transcript variants exist: e13a2 (b2a2) and e14a2 (b3a2), both encoding p210.
- The p190 isoform (from minor BCR breakpoint) is rare in CML but common in Ph+ ALL.
- There is no BCR-ABL1-negative CML - diseases formerly called this are distinct entities (atypical CML, chronic neutrophilic leukemia).
(Robbins, Cotran & Kumar Pathologic Basis of Disease, p. 583; Harrison's Principles of Internal Medicine 22E, p. 878)
Epidemiology
| Parameter | Data |
|---|---|
| Proportion of all leukemias | ~15% |
| Annual incidence (US) | ~9,000 cases/year; 2 per 100,000 |
| Worldwide annual incidence | ~250,000 cases |
| Median age at diagnosis | 55-65 years |
| Male:female ratio | 1.6:1 |
| Pediatric cases | Only 3% are <20 years |
With TKI therapy reducing annual mortality from 10-20% to ~1-2%, CML prevalence is projected to plateau at ~450,000 in the US by 2040.
Risk factors are minimal. Ionizing radiation (nuclear accidents, high-dose radiation therapy) increases risk, peaking 5-10 years post-exposure. No associations with benzene, viruses, or familial clustering.
(Harrison's 22E, p. 878)
Disease Phases
CML follows a biphasic or triphasic natural history. Phase definitions differ slightly between the WHO and European Leukemia Network (ELN) classifications:
Chronic Phase (CP)
-
75% of CML diagnoses are made here
- Indolent, dominated by granulocytic proliferation
- Peripheral blood blasts <10% (WHO) / <15% (ELN)
- Responds well to TKI therapy
Accelerated Phase (AP)
- Increasing resistance to therapy
- ELN criteria: peripheral/BM blasts 15-30%; additional cytogenetic clones (e.g., trisomy 8, isochromosome 17q, Ph duplication)
- WHO criteria: blasts 10-20%
- Increasing anemia, thrombocytopenia, rising basophil count
Blast Phase / Blast Crisis (BP)
- ELN: blasts ≥30%; WHO: blasts ≥20%
- Behaves like acute leukemia
- Myeloid blast crisis in ~70% of cases
- Lymphoid (pre-B cell) blast crisis in ~25-30%
- The lymphoid subtype implicates IKZF1 (Ikaros) mutations in ~85% of cases, same mutations seen in BCR-ABL+ B-ALL
Without treatment: ~50% progress through the accelerated phase over ~3 years; the other 50% transform directly to blast crisis.
(Goldman-Cecil Medicine, p. 1938; Robbins Pathology, p. 583-584)
Morphology
Peripheral Blood:
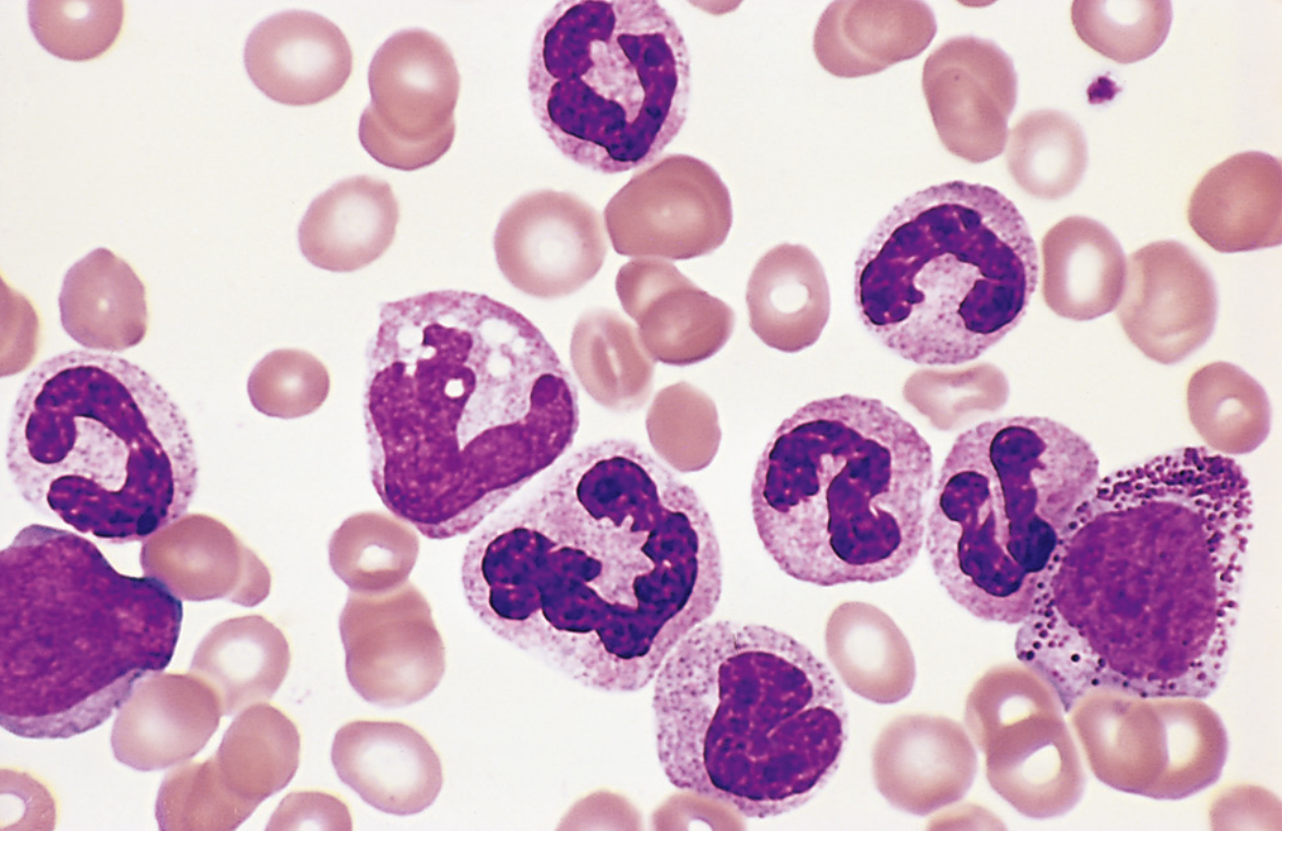
- Leukocytosis often exceeding 100,000 cells/μL, predominantly neutrophils, band forms, metamyelocytes, myelocytes
- Elevated eosinophils and basophils
- Thrombocytosis (often marked)
- Blasts <10% in chronic phase
- Low leukocyte alkaline phosphatase (LAP) score - a classic finding
Bone Marrow:
- Markedly hypercellular with massively increased maturing granulocytic precursors
- Increased small, dysplastic megakaryocytes
- Erythroid progenitors normal or mildly decreased
- Sea-blue histiocytes (macrophages with wrinkled greenish cytoplasm) - a characteristic finding
- Increased reticulin; overt fibrosis is rare in chronic phase
Spleen:
- Often massively enlarged due to extramedullary hematopoiesis (can exceed 2,600 g vs. normal ~150-200 g)
- Contains infarcts of varying age
(Robbins Pathology, p. 583)
Clinical Features
- Onset is insidious; many patients are diagnosed incidentally on routine CBC
- Constitutional symptoms from hypermetabolism: fatigue, weakness, weight loss, anorexia, night sweats
- Splenomegaly - often massive; can cause dragging sensation in left upper quadrant or acute pain from splenic infarction
- Mild-to-moderate anemia
- Rarely: hyperviscosity symptoms, gout (urate overproduction), priapism
Differentiation from other myeloproliferative neoplasms rests on detection of BCR::ABL1 by cytogenetics, FISH, or PCR - not on morphology alone.
Diagnosis
A stepwise approach is required:
- CBC + differential - leukocytosis with left shift, basophilia, thrombocytosis
- Peripheral blood smear - granulocyte spectrum, basophils
- Bone marrow biopsy/aspirate - cellularity, blast%, fibrosis
- Cytogenetics (karyotype) - identify Ph chromosome t(9;22)
- FISH - detects Ph-negative BCR::ABL1-positive cases
- Quantitative RT-PCR (qRT-PCR) - quantifies BCR::ABL1 transcript level on the International Scale (IS); baseline and for monitoring response
Treatment
Tyrosine Kinase Inhibitors (TKIs) - The Cornerstone
Six FDA-approved oral BCR::ABL1 TKIs exist:
| Agent | Generation | Dose (frontline) | Notable Toxicities |
|---|---|---|---|
| Imatinib (Gleevec) | 1st | 400 mg daily | Edema, nausea, musculoskeletal pain, rash |
| Dasatinib (Sprycel) | 2nd | 100 mg daily | Pleural/pericardial effusions, pulmonary hypertension, myelosuppression |
| Nilotinib (Tasigna) | 2nd | 300 mg twice daily | Diabetes, arterio-occlusive events, pancreatitis |
| Bosutinib (Bosulif) | 2nd | 400 mg daily | Diarrhea, liver toxicity, renal dysfunction |
| Ponatinib (Iclusig) | 3rd | 45 mg daily (reduce to 15 mg once CCyR) | Arterio-occlusive events (10-20%), hypertension, pancreatitis |
| Asciminib (Scemblix) | 3rd / STAMP inhibitor | 40 mg twice daily | Arterial occlusive events, hypertension |
Key points:
- Nilotinib is structurally similar to imatinib but 30x more potent
- Dasatinib is 300x more potent than imatinib and also inhibits SRC kinases
- Bosutinib is 30-50x more potent than imatinib and also inhibits SRC kinases
- Ponatinib and asciminib are indicated for T315I-mutated CML (the "gatekeeper" mutation that confers resistance to all 1st/2nd generation TKIs)
- Asciminib (STAMP inhibitor) binds the ABL myristoyl pocket - a completely different binding site from all other TKIs, making it uniquely active against many resistance mutations
- Omacetaxine (Synribo) - a non-TKI option (protein synthesis inhibitor) for patients failing ≥2 TKIs
(Harrison's 22E, pp. 882-883)
Monitoring and Response Milestones
Response is monitored by qRT-PCR (BCR::ABL1 IS) and cytogenetics. Key definitions:
| Response | Definition |
|---|---|
| Complete Hematologic Response (CHR) | Normal CBC, no symptoms, spleen normal |
| Complete Cytogenetic Response (CCyR) | 0% Ph+ metaphases on karyotype |
| Major Molecular Response (MMR / MR3.0) | BCR::ABL1 IS ≤0.1% (3-log reduction) |
| MR4.0 / Deep Molecular Response | BCR::ABL1 IS ≤0.01% |
| MR4.5 | BCR::ABL1 IS ≤0.0032% |
ELN and NCCN define "failure" and "warning" milestones at 3, 6, 12 months - guiding when to switch TKIs.
Treatment-Free Remission (TFR)
A major emerging concept: patients achieving sustained deep molecular response (MR4.0 or MR4.5 for ≥2 years) may attempt TKI discontinuation. Approximately 40-60% maintain remission off therapy long-term. Monthly PCR monitoring is mandatory after stopping; ~50% who lose MMR regain it after restarting the same TKI.
Allogeneic HSCT
Previously the only curative option, now reserved for:
- Blast phase or accelerated phase
- TKI resistance/failure after multiple lines
- T315I mutation not responsive to ponatinib/asciminib
- Healthy, younger patients with high-risk disease
ABL1 Kinase Domain Mutations and Resistance
When patients fail TKI therapy, ~50% of cases are due to point mutations in the ABL1 kinase domain preventing TKI binding (e.g., T315I). Testing for mutations guides selection of the next TKI. The other ~50% involve BCR::ABL1-independent resistance mechanisms (other kinase mutations, BCR::ABL1 amplification, clonal evolution).
Prognosis
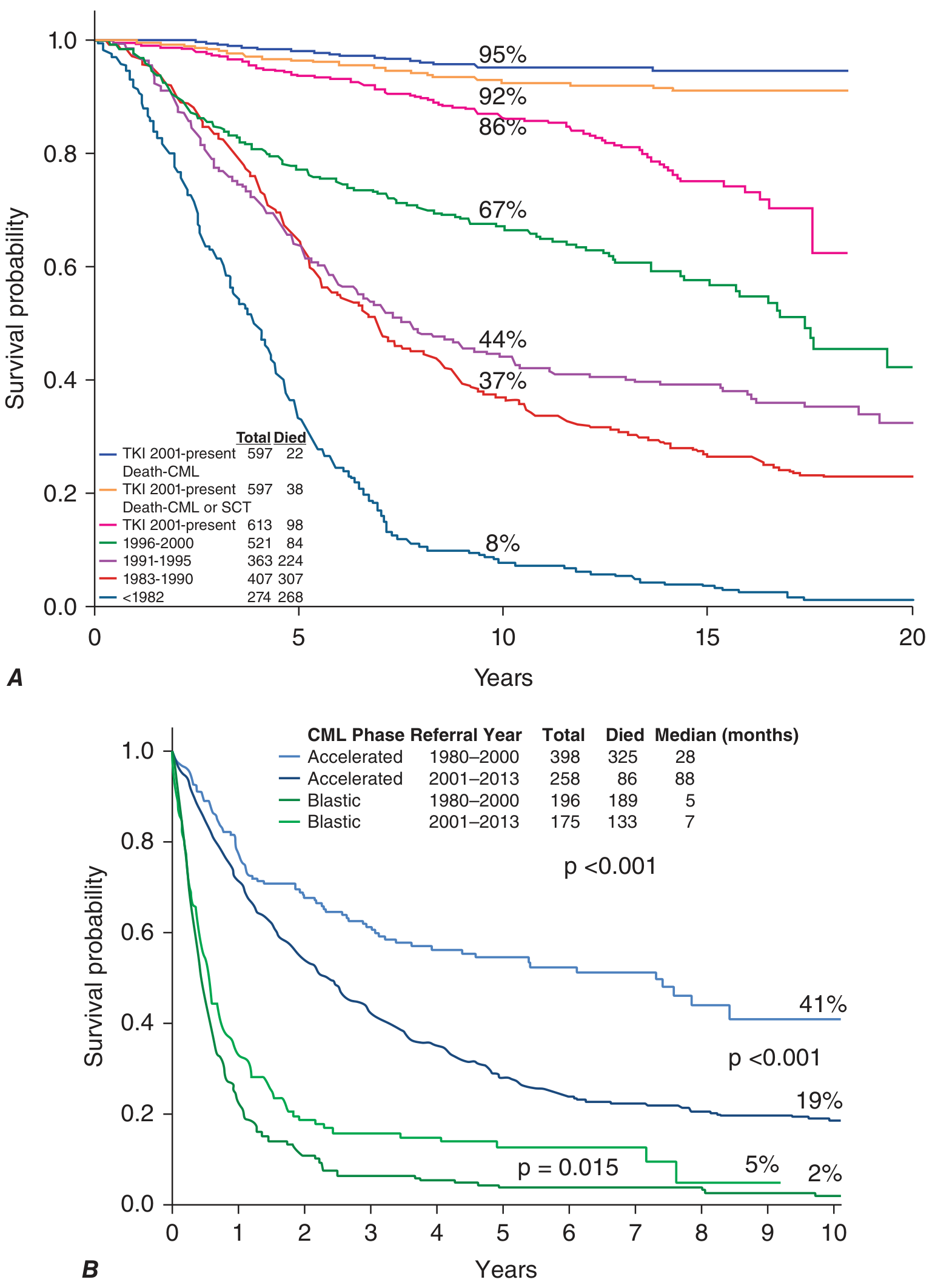
- Chronic phase with TKIs: 10-year survival >85%, approaching normal life expectancy. In the TKI era (2001-present), only 22 out of 597 patients died from CML-related causes at 10 years (survival ~95% for CML-specific death).
- Accelerated phase with TKIs: Median survival improved from 28 months (pre-TKI era) to 88 months.
- Blast crisis: Remains very poor; median survival only 5-7 months even in the TKI era.
Prognostic scoring systems (Sokal, Hasford/Euro, EUTOS, ELTS) stratify patients at diagnosis into low/intermediate/high risk, though their relevance has diminished with effective TKI therapy.
Key Distinguishing Points
| Feature | CML |
|---|---|
| Genetic lesion | BCR::ABL1 (Ph chromosome), 100% of cases |
| Oncoprotein | p210 BCR-ABL (constitutive tyrosine kinase) |
| LAP score | Low (distinguishes from leukemoid reaction) |
| Basophilia | Characteristic; basophil count predicts transformation |
| Targeted therapy | BCR::ABL1 TKIs - imatinib, nilotinib, dasatinib, bosutinib, ponatinib, asciminib |
| Blast crisis type | Myeloid 70%, lymphoid ~25-30% |
| Curative option | Allogeneic HSCT (now late-line only) |
Sources: Robbins, Cotran & Kumar Pathologic Basis of Disease pp. 582-585 | Harrison's Principles of Internal Medicine 22E pp. 878-884 | Goldman-Cecil Medicine pp. 1938-1940
Recent evidence note: A 2024 systematic review (PMID 39634261) comparing ponatinib vs. asciminib after 2nd-generation TKI failure found broadly comparable outcomes, supporting asciminib as a viable option in this setting - consistent with current guidelines. A 2025 meta-analysis (PMID 40317312) systematically documented patient-reported TKI toxicity symptoms, reinforcing the clinical importance of TKI side effect monitoring and dose optimization.
This is a shared conversation. Sign in to Orris to start your own chat.