Explain the mechanism of acute respiratory distress syndrome
Reading File
Finding Sources
Finding Sources
Searching PubMed
"acute respiratory distress syndrome"[MeSH Terms] AND pathophysiology
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Loading Image
I now have all the content I need from multiple authoritative sources. Let me compile a comprehensive, well-structured answer.
Mechanism of Acute Respiratory Distress Syndrome (ARDS)
ARDS is a clinical syndrome of acute respiratory failure caused by diffuse alveolar damage (DAD) - a non-cardiogenic, protein-rich pulmonary edema resulting from disruption of the alveolar-capillary barrier. It carries a mortality rate commonly exceeding 30%.
Berlin Definition (2012)
| Criterion | Definition |
|---|---|
| Timing | Within 1 week of a known precipitant |
| Imaging | Bilateral opacities on CXR/CT not explained by effusions or collapse |
| Origin of edema | Not explained by cardiac failure or fluid overload |
| Oxygenation (Mild) | PaO2/FiO2 200-300 mmHg with PEEP ≥5 cm H2O |
| Oxygenation (Moderate) | PaO2/FiO2 100-200 mmHg with PEEP ≥5 cm H2O |
| Oxygenation (Severe) | PaO2/FiO2 <100 mmHg with PEEP ≥5 cm H2O |
Precipitating Causes
Risk factors are divided by mechanism of injury:
| Direct (Pulmonary) Injury | Indirect (Non-pulmonary) Injury |
|---|---|
| Pneumonia (bacterial, viral, COVID-19) | Sepsis |
| Aspiration of gastric contents | Major trauma |
| Pulmonary contusion | Multiple blood transfusions |
| Toxic inhalation / smoke | Pancreatitis |
| Near-drowning | Cardiopulmonary bypass |
| Reperfusion injury (post-lung transplant) | Drug overdose |
Sepsis, trauma, aspiration, and massive transfusion carry the highest risk. Chronic alcoholism doubles ARDS risk and worsens mortality.
Phases of ARDS
The natural history follows three overlapping phases:

Core Pathophysiology: The Alveolar-Capillary Barrier
The central event in ARDS is breakdown of the alveolar-capillary barrier, which normally consists of the capillary endothelium, the interstitium, and the alveolar epithelium (Type I and Type II pneumocytes). When this barrier fails, protein-rich fluid floods the alveoli, producing non-hydrostatic (exudative) pulmonary edema - distinguishing ARDS from cardiogenic edema where hydrostatic pressure is the driver.
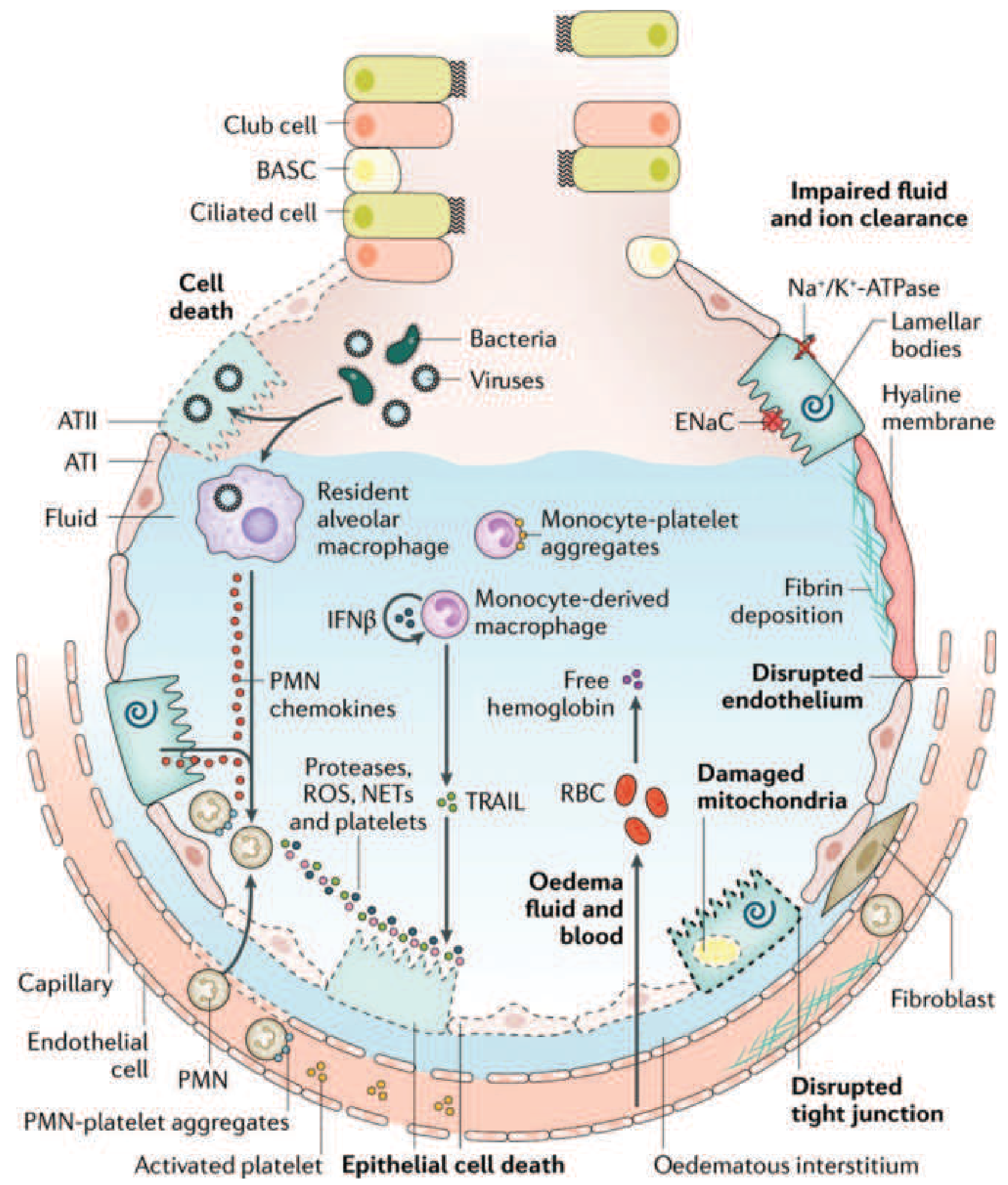
Phase 1: Exudative Phase (Days 0-7)
1. Initiation - Endothelial and Epithelial Injury
The initiating insult (direct or indirect) activates Toll-like receptors (TLRs) on alveolar Type I (ATI) epithelial cells and resident alveolar macrophages. This triggers downstream inflammatory cascades including NF-κB signaling, releasing pro-inflammatory cytokines: TNF-α, IL-1β, IL-6, IL-8, and others.
- Endothelial injury: disrupts tight junctions between capillary endothelial cells, increasing vascular permeability
- Epithelial injury: Type I pneumocytes (which cover 95% of the alveolar surface) are particularly vulnerable; their necrosis disrupts the epithelial barrier
2. Neutrophil Recruitment and Activation
One of the histological hallmarks of ARDS is accumulation of neutrophils in the pulmonary microvasculature - neutrophils are central to its pathogenesis.
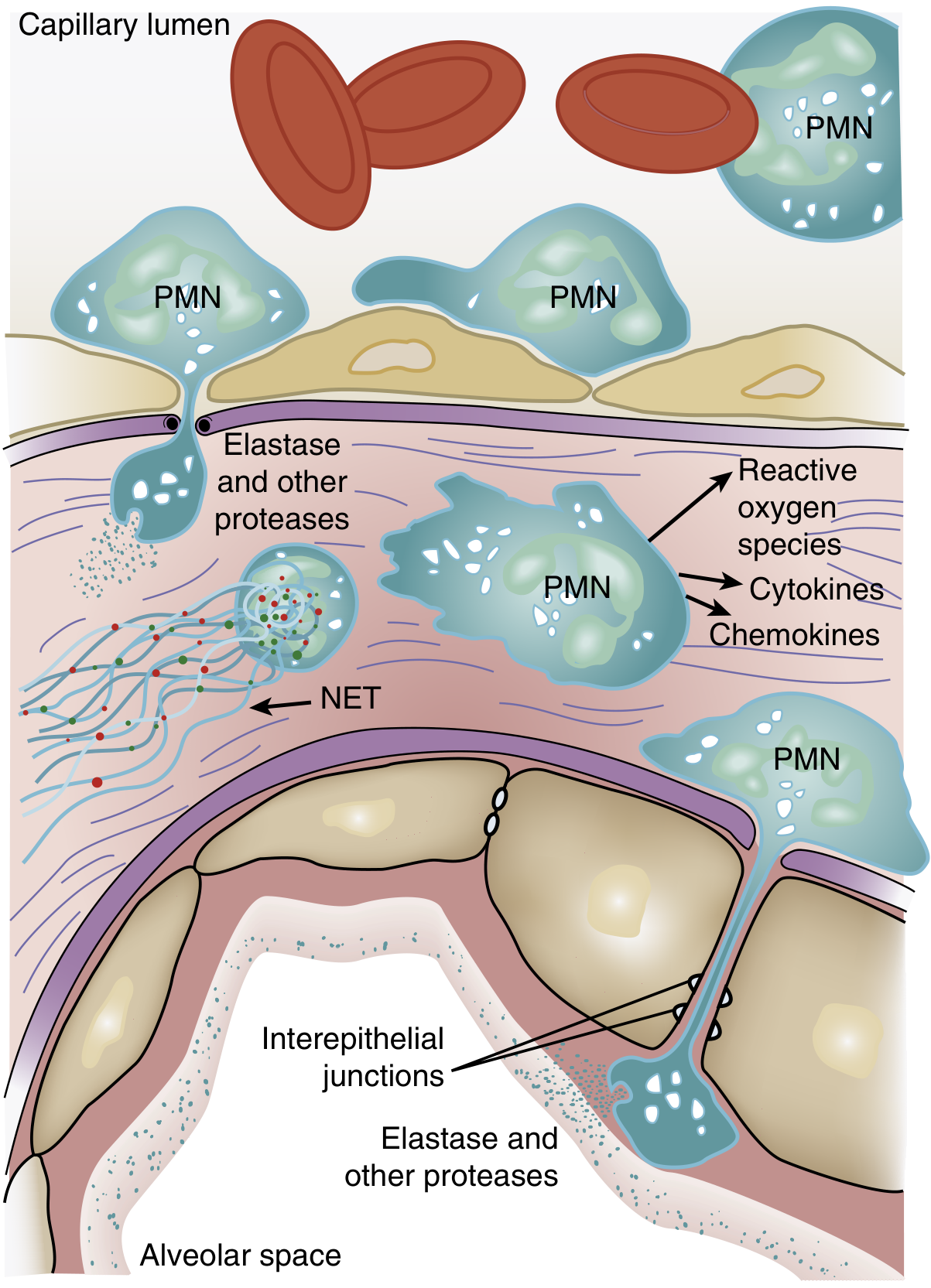
The sequence:
- Transient leukopenia is often the earliest manifestation of ARDS - neutrophils sequester in the pulmonary capillaries even before hypoxemia develops
- Activated neutrophils become mechanically "stiff" (via actin cytoskeleton remodeling) and cannot deform to pass through narrow pulmonary capillary segments (average capillary diameter < average neutrophil diameter)
- Sequestered neutrophils migrate across the alveolar-capillary membrane - this can occur even without conventional adhesion molecules (L-selectin, β2-integrins)
- Once in the alveolar space, activated neutrophils release a destructive arsenal:
- Reactive oxygen species (ROS) - oxidative injury to membranes
- Proteolytic enzymes - leukocyte elastase, matrix metalloproteinases
- Cationic peptides (defensins)
- Eicosanoids
- Neutrophil extracellular traps (NETs) - chromatin-based networks that trap pathogens but also damage host tissue
- Cytokines/chemokines - amplify the inflammatory response (TNF-α, IL-1β)
IL-8 is a particularly potent neutrophil chemokine; its BAL concentration is elevated in ARDS patients and correlates with severity. The presence of neutrophils in BAL fluid correlates with a poor prognosis.
3. Alveolar Flooding and Edema Formation
As a result of barrier disruption:
- Protein-rich, exudative fluid floods the alveolar spaces
- This edema fluid contains fibrin, inflammatory cells, and plasma proteins
- Hypoxemia develops from ventilation-perfusion (V/Q) mismatch and intrapulmonary right-to-left shunting (blood perfuses non-ventilated, fluid-filled alveoli)
- Dead space increases substantially (sometimes to >60% of tidal volume), impairing CO2 elimination
4. Surfactant Dysfunction
- Alveolar flooding dilutes and inactivates surfactant
- Inflammatory mediators (especially phospholipase A2, elevated in pancreatitis-associated ARDS) enzymatically degrade surfactant phospholipids
- Loss of surfactant leads to increased alveolar surface tension, promoting alveolar collapse (atelectasis) and further worsening compliance and shunting
- Surfactant proteins (SP-A, SP-D) also have antimicrobial roles; their loss may impair host defense
5. Hyaline Membrane Formation
The protein-rich exudate that floods alveoli coagulates along denuded alveolar walls, forming hyaline membranes - the pathologic hallmark of DAD on biopsy. These membranes are composed of fibrin, plasma proteins, and cellular debris lining the alveolar walls.
6. Coagulation Activation
The ARDS alveolus is strongly procoagulant:
- Tissue factor is expressed on injured epithelial cells
- Fibrin deposition in alveoli and pulmonary microvasculature impairs gas exchange
- Microvascular thrombosis reduces pulmonary blood flow and contributes to pulmonary hypertension
Phase 2: Proliferative Phase (Days 7-21)
If the patient survives the exudative phase, a reparative process begins:
- The neutrophil-predominant infiltrate shifts to a lymphocyte-predominant one
- Type II pneumocytes (which survived the initial injury better than Type I cells) proliferate along denuded alveolar basement membranes
- Type II cells synthesize new surfactant and differentiate into Type I pneumocytes to restore the epithelial lining
- Alveolar exudates begin to organize
- Many patients recover during this phase; others develop progressive injury and early fibrotic changes
Phase 3: Fibrotic Phase (Days 21+)
A subset of patients progresses to irreversible fibrosis:
- Alveolar edema and inflammatory exudates are replaced by alveolar-duct and interstitial fibrosis
- Marked disruption of acinar architecture leads to emphysema-like changes with large bullae
- Intimal fibroproliferation in pulmonary microcirculation causes progressive vascular occlusion and pulmonary hypertension
- Physiologic consequences: increased pneumothorax risk, reduced compliance, increased dead space
- Lung biopsy evidence of fibrosis in any ARDS phase is associated with increased mortality
Physiologic Consequences of the Mechanism
| Mechanism | Physiologic Effect |
|---|---|
| Alveolar flooding + atelectasis | Intrapulmonary shunt → refractory hypoxemia |
| V/Q mismatch | Hypoxemia |
| Increased dead space ventilation | Hypercapnia, increased work of breathing |
| Reduced lung compliance | Higher airway pressures needed for ventilation |
| Pulmonary vasoconstriction (hypoxic) | Pulmonary hypertension, RV strain |
| Surfactant loss | Alveolar collapse, reduced compliance |
Ventilator-Induced Lung Injury (VILI) - A Superimposed Mechanism
Because ARDS lung is heterogeneous (consolidated dependent zones interspersed with relatively normal non-dependent zones), mechanical ventilation itself can amplify injury through several mechanisms:
- Volutrauma: large tidal volumes preferentially overdistend the "baby lung" (the small fraction of normal-appearing alveoli), causing mechanical injury and increased permeability
- Barotrauma: high airway pressures cause pneumothorax, pneumomediastinum
- Atelectrauma: repetitive opening and closing of collapsed airway units generates high shear stresses at the interface of aerated and collapsed tissue
- Biotrauma: mechanical stretch triggers local cytokine release (TNF-α, IL-1β, IL-6, IL-8) and systemic inflammation, contributing to multi-organ dysfunction
This is the rationale for lung-protective ventilation (6 mL/kg tidal volume, plateau pressure ≤30 cm H2O, PEEP to prevent derecruitment).
Genetic and Host Factors
Several genetic variants modify susceptibility to ARDS. Notably, SARS-CoV-1 infection via the ACE2 receptor reduces ACE2 levels, thereby promoting acute lung injury - a mechanism relevant to COVID-19-associated ARDS as well. Alcoholism doubles ARDS risk through unclear mechanisms, possibly involving impaired alveolar epithelial defense and oxidant stress.
Summary Diagram of the Mechanism
Insult (sepsis, pneumonia, trauma, aspiration...)
↓
Alveolar macrophage + TLR activation
↓
Cytokine storm (TNF-α, IL-1β, IL-6, IL-8)
↓
Neutrophil sequestration & transmigration into alveoli
↓
Release of ROS, proteases, NETs
↓
Endothelial + Epithelial barrier disruption
↓
Protein-rich exudate floods alveoli
Surfactant degradation + hyaline membrane formation
Microvascular fibrin deposition
↓
Shunt + V/Q mismatch + dead space → Hypoxemia
Reduced compliance → Increased work of breathing
Pulmonary hypertension → RV dysfunction
↓
↙ Recovery (Type II cell proliferation, repair)
↘ Fibrosis (progressive vascular occlusion, bullae)
Sources:
- Murray & Nadel's Textbook of Respiratory Medicine, Chapter 134 (ARDS - Etiology, Pathogenesis, Neutrophil Biology, VILI)
- Harrison's Principles of Internal Medicine, 22E, Chapter 312 (Clinical Course and Pathophysiology)
- Goldman-Cecil Medicine, Chapter 91 (Ventilatory Support and Lung-Protective Strategies)
- Sabiston Textbook of Surgery, Chapter on ARDS (Berlin Definition, Management)
- Robbins & Kumar Basic Pathology (Diffuse Alveolar Damage, Histopathology)
This is a shared conversation. Sign in to Orris to start your own chat.