Amyloidosis
Reading File
Finding Sources
Searching PubMed
amyloidosis treatment
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Reading File
Reading File
Reading File
I now have comprehensive information from multiple authoritative textbooks. Let me compile a complete, well-structured answer.
Amyloidosis
Amyloidosis describes a heterogeneous group of disorders characterized by extracellular deposition of misfolded proteins as insoluble fibrils that disrupt tissue architecture and cause organ dysfunction. More than 30 different precursor proteins can form amyloid in humans; 13 can cause systemic disease.
Structural Biology
All amyloid deposits share a remarkably uniform morphology regardless of their precursor protein:
- Rigid, non-branching fibrils assembled on cross-beta scaffolds, reinforced by steric zipper motifs forming a stable beta-pleated sheet configuration
- Associated non-fibrillar components: serum amyloid P (SAP), apolipoprotein E, glycosaminoglycans (heparan and dermatan sulfate groups), laminin, and collagen IV
- On H&E stain: homogeneous, hyaline-like, eosinophilic extracellular material
- On Congo red stain: pink/red in normal light; apple-green birefringence under polarized light - the gold standard diagnostic finding
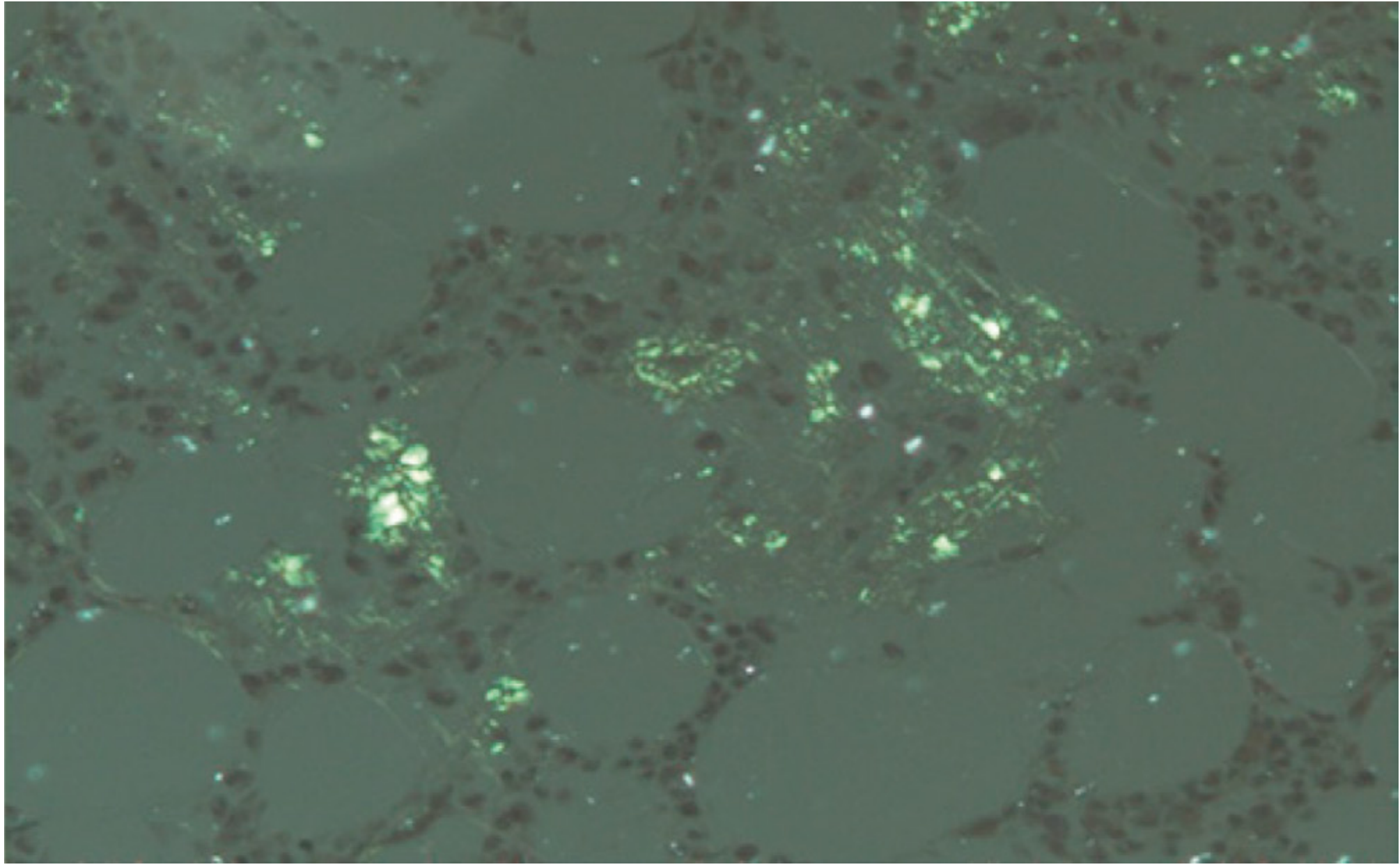
Other staining characteristics: weakly PAS-positive, diastase resistant, purple with crystal violet, positive with thioflavin T.
Pathogenesis
Organ dysfunction arises primarily from the abnormal tissue architecture created by deposits. Some amyloidogenic proteins additionally activate signaling pathways that produce ROS and impair calcium homeostasis.
Normally, misfolded proteins are degraded intracellularly by proteasomes or extracellularly by macrophages. In amyloidosis, these quality-control mechanisms fail. Amyloidogenic proteins fall into two broad categories:
- Normal proteins produced in excess quantities that have an inherent tendency to misfold
- Variant (mutant) proteins that are intrinsically prone to misfolding even at normal concentrations
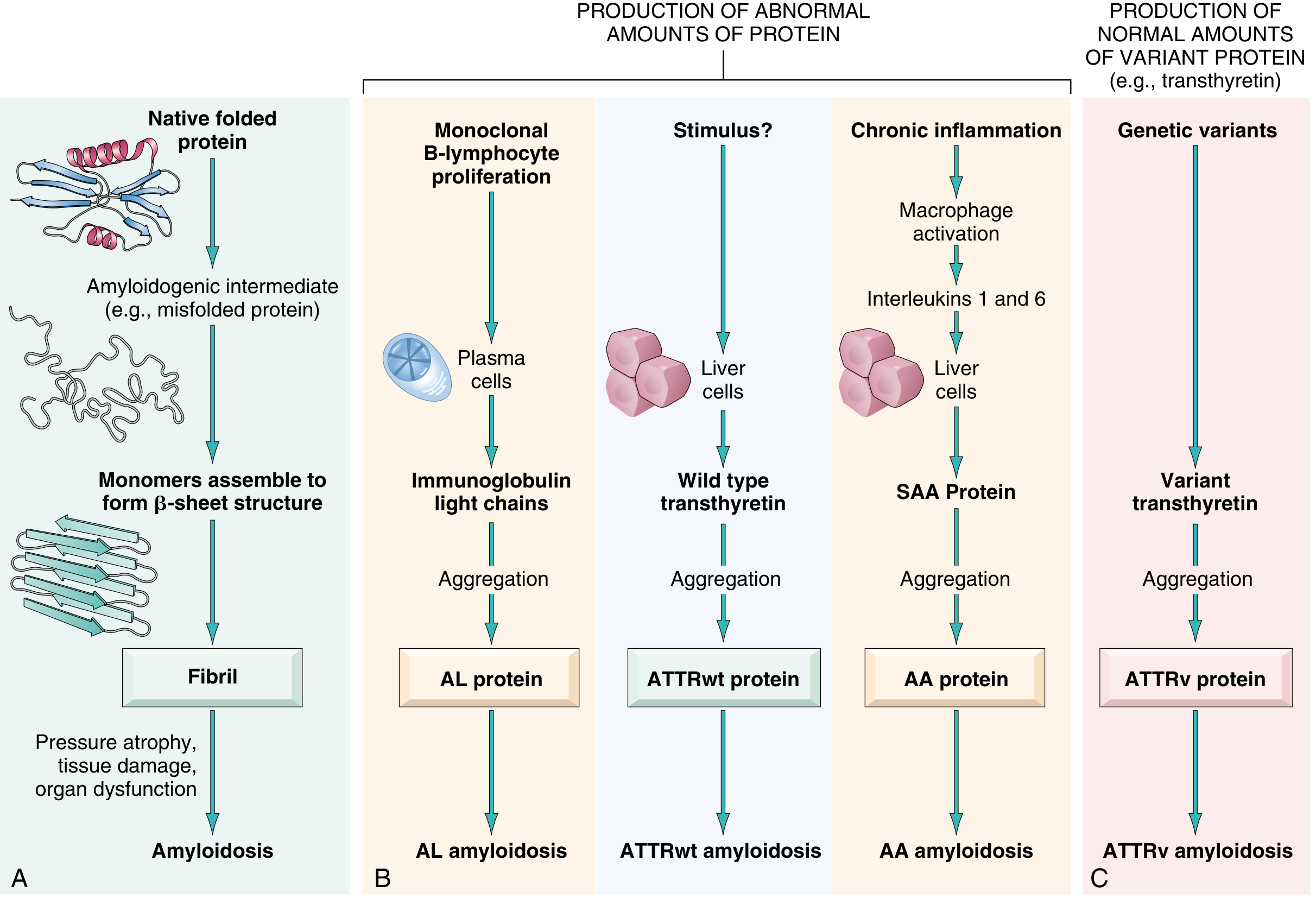
Classification
| Type | Fibril Protein | Precursor | Associated Condition |
|---|---|---|---|
| AL (primary) | Immunoglobulin light chain (chiefly lambda) | Monoclonal plasma cell proliferation | Multiple myeloma, MGUS |
| AA (secondary) | Serum amyloid A (SAA) | Chronic inflammation - macrophages secrete IL-1/IL-6, driving hepatic SAA | Rheumatoid arthritis, Crohn's, FMF, TB |
| ATTRwt | Wild-type transthyretin | Liver; age-related destabilization | Elderly males (>70 yrs) |
| ATTRv | Variant transthyretin | Liver; >130 mutations, chromosome 18 | Familial amyloid polyneuropathy/cardiomyopathy |
| Abeta | Beta-amyloid from APP cleavage | Neuronal/vascular | Alzheimer disease |
| Abeta2m | beta-2-microglobulin (MHC class I component) | Retained in renal failure | Long-term hemodialysis |
| ACal | Calcitonin precursor | C cells | Medullary thyroid carcinoma |
| AIAPP | Islet amyloid polypeptide | Pancreatic beta cells | Type 2 diabetes |
| AA | SAA | Autoinflammatory/IL-1 excess | Familial Mediterranean fever |
Major Clinical Types in Detail
AL Amyloidosis (most common systemic form - ~2,000-3,000 new US cases/year)
- Caused by clonal plasma cells synthesizing amyloidogenic immunoglobulin light chains (lambda > kappa)
- Occurs in 5-15% of multiple myeloma patients; also with MGUS
- Organ targets: heart (50-75%), kidneys (nephrotic syndrome), peripheral nerves, GI tract, liver, tongue (macroglossia), skin, respiratory tract
- Untreated median survival of AL cardiomyopathy: 1.5 years
AA Amyloidosis (secondary)
- Complicates chronic inflammatory/infectious diseases: RA, Crohn's, bronchiectasis, tuberculosis, osteomyelitis, FMF
- Mechanism: chronic inflammation → macrophage activation → IL-1/IL-6 → hepatic SAA overproduction
- Primary target: kidneys (proteinuria, nephrotic syndrome, eventually ESRD)
- Treated by controlling the underlying inflammatory disease; colchicine effective in FMF-related AA
- Potassium permanganate pretreatment abolishes Congo red staining of AA but not AL amyloid - useful for distinguishing the two histologically
ATTR Amyloidosis
Wild-type (ATTRwt): Age-related destabilization of the transthyretin tetramer (a liver-derived protein that carries <5% thyroxine and retinol-binding protein). Affects males >70 years primarily with restrictive cardiomyopathy. Untreated median survival: 3.6 years.
Variant (ATTRv): >130 mutations; most common in the US is Val122Ile (p.V142I) - present in 3.4% of Black Americans and common in those of African Caribbean descent. Causes cardiomyopathy and/or peripheral/autonomic neuropathy. Median survival untreated: 2.5 years.
Morphology / Organ Changes
Kidney: Deposits begin in mesangium and glomerular basement membrane → nephrotic syndrome → renal failure. Cortex appears enlarged and pale/waxy.
Heart: LV wall thickening, restrictive cardiomyopathy, HFpEF in early stages (HFrEF late). Cardiac amyloidosis particularly involves AL and ATTR types.
Liver: Amyloid deposits in space of Disse → hepatomegaly with "waxy liver" appearance. Liver function usually preserved early.
Spleen: "Sago spleen" (deposits in follicles) or "lardaceous spleen" (diffuse). Splenomegaly.
Nervous system: Peripheral neuropathy (especially vATTR), autonomic neuropathy, carpal tunnel syndrome (often a sentinel symptom of ATTRwt).
Diagnosis
Histology remains the gold standard: Congo red staining with apple-green birefringence under polarized light.
- Sensitivity depends on amyloid quantity, correct tissue processing, and observer experience
- A negative result does not exclude amyloidosis (patchy distribution)
- Biopsy sites (in order of preference/yield): abdominal fat pad aspiration (sensitivity varies - only 45% for vATTR, 15% for wtATTR), rectal biopsy, minor salivary gland, affected organ (renal/endomyocardial biopsy)
- Subtyping is mandatory after amyloid detection: immunohistochemistry (IHC), immunofluorescence, or - most accurately - laser microdissection/mass spectrometry proteomics
Cardiac-specific workup:
- Echocardiography: concentric LV wall thickening (≥1.2 cm), granular/scintillating myocardial texture, biatrial enlargement, diastolic dysfunction, relative apical sparing pattern on global longitudinal strain (GLS) - hallmark finding
- Cardiac MRI: late gadolinium enhancement in a diffuse, subendocardial/transmural pattern; T1 mapping elevated; extracellular volume (ECV) increased
- Tc-99m pyrophosphate (PYP) / DPD / HMDP scintigraphy: Grade 2-3 uptake is highly specific for ATTR cardiomyopathy (>99% specificity when M-protein excluded); allows non-invasive diagnosis of ATTR without biopsy
- ECG: low voltage (especially in AL), pseudo-infarct pattern, conduction abnormalities
Serum/urine tests: Serum free light chains, SPEP/UPEP (for AL); genetic testing for TTR mutations (all ATTR patients); SAA levels (for AA monitoring).
Treatment
AL Amyloidosis
The target is eliminating the clonal plasma cell source of amyloidogenic light chains:
| Approach | Regimen | Notes |
|---|---|---|
| Autologous stem cell transplant (ASCT) | Myeloablative conditioning | Eligible patients only; transplant mortality ~2.5% at expert centers; 10-yr survival 53% if complete response |
| Standard chemotherapy | CyBorD (cyclophosphamide + bortezomib + dexamethasone) | Hematologic response 60%, cardiac response 17% |
| Daratumumab-based | Daratumumab + CyBorD (ANDROMEDA trial) | Complete response 53.3% vs 18.1% with CyBorD alone; now preferred first-line for transplant-ineligible |
ATTR Amyloidosis
Three mechanistic strategies:
-
TTR tetramer stabilization: Tafamidis - the only FDA-approved drug for ATTR cardiomyopathy in the US; binds TTR's thyroxine-binding sites to prevent tetramer dissociation. ATTR-ACT trial demonstrated reduced mortality and CV hospitalizations.
-
TTR silencing (RNA-based therapies):
- Patisiran (siRNA) - FDA-approved for vATTR polyneuropathy; shown to reduce cardiac troponin T and NTproBNP
- Inotersen (antisense oligonucleotide) - FDA-approved for vATTR polyneuropathy
- Vutrisiran (siRNA) - approved for vATTR polyneuropathy; HELIOS-B trial data emerging for cardiomyopathy
- Eplontersen - emerging data
-
Fibril disruption/clearance: Investigational agents (doxycycline/tauroursodeoxycholic acid combinations, anti-fibril antibodies)
AA Amyloidosis
- Treat the underlying disease - suppress SAA production
- Colchicine for FMF (prevents attacks and SAA rises)
- Biologic therapy (IL-1 or IL-6 inhibitors) for refractory inflammatory disease
- Renal transplantation for ESRD (14% risk of amyloid recurrence in the graft)
Supportive / Heart Failure Management
- Loop diuretics for fluid overload
- Avoid digoxin (binds amyloid fibrils, can cause toxicity), calcium channel blockers (negative inotropy, may worsen), and high-dose ACE inhibitors/ARBs in hypotension
- ICD for secondary prevention in carefully selected patients with expected survival >1 year
Recent Evidence
A 2025 systematic review on ATTR amyloidosis epidemiology (PMID 39819351) highlights that ATTRwt is substantially underdiagnosed in heart failure populations, with prevalence estimates as high as 13-17% in HFpEF with LV hypertrophy. This is driving increasingly aggressive screening efforts.
Sources: Robbins, Cotran & Kumar Pathologic Basis of Disease; Fuster & Hurst's The Heart, 15th Edition; Firestein & Kelley's Textbook of Rheumatology; Andrews' Diseases of the Skin; Brenner & Rector's The Kidney
This is a shared conversation. Sign in to Orris to start your own chat.