Copper metabolism
Reading File
Finding Sources
Finding Sources
Searching PubMed
"copper metabolism" homeostasis
Reading File
Reading File
Finding Sources
Reading File
Reading File
Loading Image
Loading Image
I now have comprehensive, well-illustrated content from multiple authoritative textbooks. Here is the full answer:
Copper Metabolism
Overview
Copper (Cu) is an essential trace element that is simultaneously indispensable for life and toxic in excess. Dietary intake typically ranges from 1.5-4 mg/day, and the body maintains tight homeostasis through coordinated absorption, transport, intracellular chaperoning, and biliary excretion.
1. Dietary Sources and Absorption
Sources: Shellfish, liver, nuts, legumes, bran, organ meats.
Absorption occurs mainly in the proximal small intestine (stomach and duodenum). At the enterocyte brush border, the copper transporter CTR1 (SLC31A1) mediates uptake of Cu²⁺ after it is reduced to Cu⁺. Inside the enterocyte, the P-type ATPase ATP7A pumps copper across the basolateral membrane into the portal circulation. This step is the site of defect in Menkes disease (X-linked; ATP7A mutation), leading to failure of copper export from intestinal cells and a state of systemic copper deficiency.
2. Plasma Transport
After absorption, copper is loosely bound to:
- Albumin (primary carrier in portal blood)
- Histidine
- α₂-macroglobulin
Portal blood flow directs most copper to the liver. A very small fraction travels to peripheral tissues (brain, eyes, and cuproenzymes).
3. Hepatic Handling - The Central Hub
The liver is the primary organ of copper regulation. Inside hepatocytes:
- Metallochaperones (low-molecular-weight proteins) receive free copper - copper is never truly "free" within a cell.
- Key intracellular chaperones direct copper to specific destinations:
- CCS → delivers Cu to superoxide dismutase (SOD1) in the cytoplasm
- COX17 → delivers Cu to cytochrome c oxidase in the mitochondria
- ATOX1 → delivers Cu to ATP7B in the trans-Golgi network (TGN)
- Metallothioneins and glutathione act as overflow buffers, binding excess copper in a non-toxic form.
ATP7B (Wilson ATPase)
The hepatocyte's key copper pump is ATP7B, a membrane-bound P₁-type ATPase (1443 amino acids, 160 kDa, encoded on chromosome 13q14). It has two principal functions:
- Incorporation of copper into apoceruloplasmin → forms holo-ceruloplasmin, secreted into blood
- Biliary excretion of copper (via lysosomes to bile canaliculi) - the main route of copper elimination
When intracellular copper rises, ATP7B traffics from the TGN toward the apical (bile canalicular) membrane to increase biliary export.
Fig. 1 - Simplified overview of copper transport pathways and genetic disorders
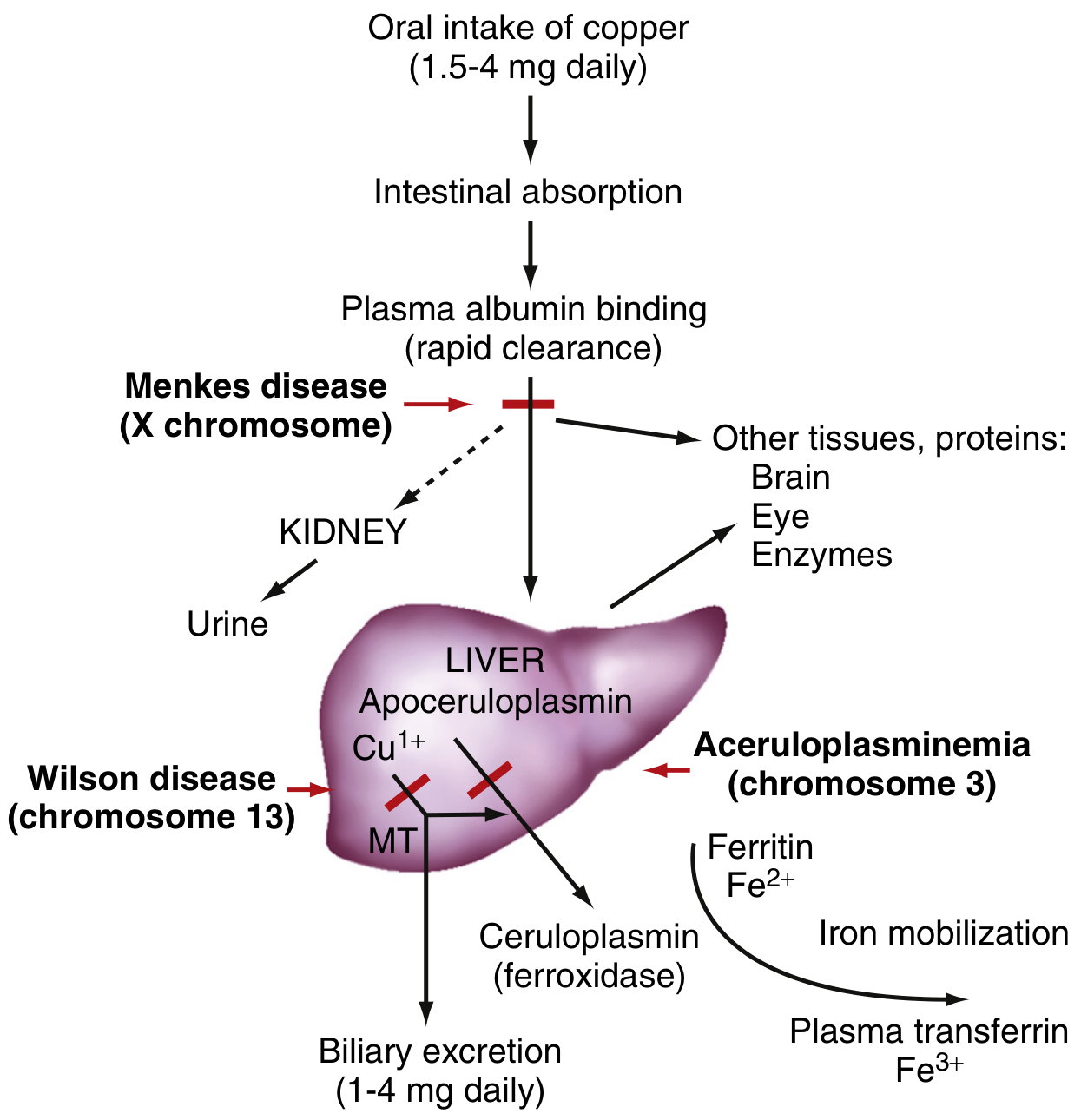
Modified from Sleisenger and Fordtran's Gastrointestinal and Liver Disease
Fig. 2 - Hepatocyte copper handling and Wilson disease mechanism
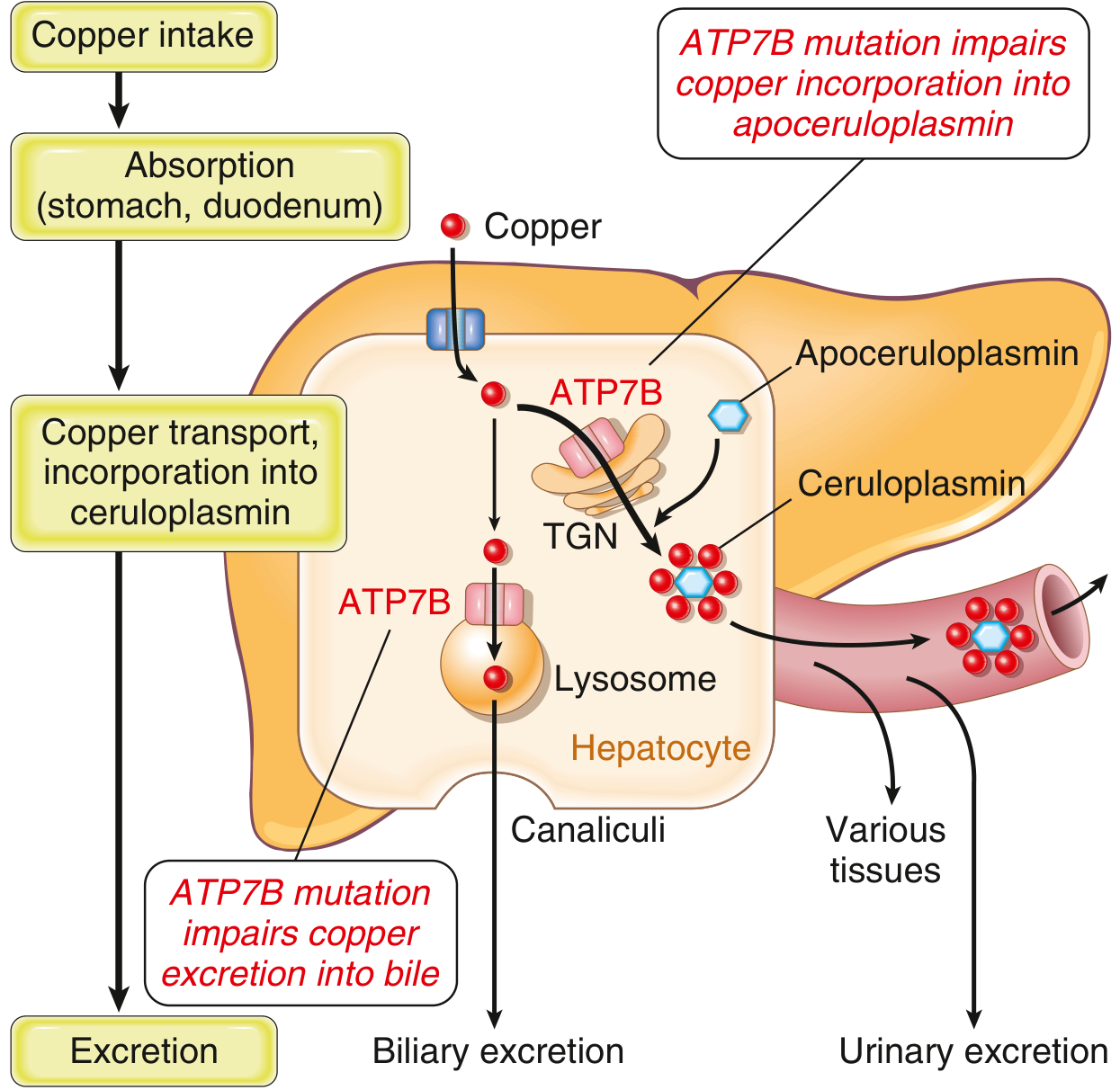
Robbins & Kumar Basic Pathology
4. Ceruloplasmin
- An α₂-glycoprotein, MW 132 kDa, containing 6 copper atoms per molecule
- Synthesized in the liver by copper loading onto apoceruloplasmin (via ATP7B)
-
90% of plasma copper is bound as an integral part of ceruloplasmin
- Normal serum level: 200-400 mg/L in adults (rises to 300-500 mg/L in early childhood, then falls)
- Acts as a ferroxidase (oxidizes Fe²⁺ → Fe³⁺ for loading onto transferrin) - hence copper deficiency can cause iron-deficiency-like anemia
- Is an acute-phase reactant: levels rise with inflammation, pregnancy, and exogenous estrogen use
5. Excretion
- Bile is the primary excretion route: 1-4 mg/day
- Urine copper excretion is normally very small (<50 µg/day)
- No regulated re-absorption mechanism exists; excess copper simply overloads the hepatic system
6. Essential Cuproenzymes
| Enzyme | Function |
|---|---|
| Cytochrome c oxidase (Complex IV) | Mitochondrial electron transport, ATP synthesis |
| Superoxide dismutase (SOD1, SOD3) | Free radical scavenging |
| Ceruloplasmin (ferroxidase) | Iron mobilization |
| Lysyl oxidase | Elastin and collagen cross-linking |
| Tyrosinase | Melanin synthesis |
| Dopamine β-monooxygenase | Catecholamine synthesis (dopamine → norepinephrine) |
| DBH / PAM | Neurotransmitter processing |
7. Disorders of Copper Metabolism
Wilson Disease (Hepatolenticular Degeneration)
- Gene: ATP7B mutation (chromosome 13q14), autosomal recessive
- Prevalence: ~1 in 30,000 (may be higher)
- Mechanism: Failure of ATP7B to export copper into bile AND to incorporate it into ceruloplasmin → copper accumulates in hepatocytes → oxidative injury → cirrhosis; overflow copper deposits in brain (basal ganglia), corneas, kidneys, bones
- Lab findings: Low serum ceruloplasmin (<20 mg/dL), low total serum copper, elevated free (non-ceruloplasmin) copper, elevated 24-hour urine copper (>100 µg/day), elevated hepatic copper (>250 µg/g dry weight - most sensitive)
- Clinical: Liver disease (acute hepatitis, chronic hepatitis, cirrhosis, acute liver failure), neuropsychiatric symptoms, Kayser-Fleischer rings (green-brown copper deposits in Descemet membrane of cornea)
- Treatment: Copper chelation (D-penicillamine, trientine), zinc supplementation (blocks intestinal absorption), liver transplant in fulminant cases
Menkes Disease (Kinky/Steely-Hair Disease)
- Gene: ATP7A mutation (X chromosome), X-linked recessive
- Mechanism: Failure of ATP7A to export copper from enterocytes → systemic copper deficiency despite normal/elevated mucosal copper
- Clinical: Intellectual disability, hypotonia, seizures, kinky/steely hair (pili torti), vascular fragility (dissecting aneurysms), death usually by age 5
- Lab: Hypocupremia, decreased ceruloplasmin
Aceruloplasminemia
- Gene: Ceruloplasmin gene (chromosome 3), autosomal recessive
- Mechanism: Loss of ferroxidase activity → iron overload in brain and viscera
- Clinical: Mental deterioration, microcytic anemia, low serum iron and copper
Copper Deficiency
- Rare in adults; occurs in: premature infants on milk diets, malabsorption syndromes, nephrotic syndrome, excess zinc supplementation (zinc competes with copper for intestinal absorption)
- Presents as refractory microcytic anemia, neutropenia, neurologic dysfunction
Copper Toxicity
- Usually accidental (ingestion of copper salts, storage in copper vessels)
- Severe cases: nausea, vomiting, hemolysis, hepatic failure, renal failure, coma
8. Copper-Iron Crosstalk
Ceruloplasmin's ferroxidase activity links copper and iron metabolism: it oxidizes Fe²⁺ to Fe³⁺, enabling loading onto transferrin for systemic iron delivery. Copper deficiency therefore impairs iron mobilization from stores, producing an anemia that resembles iron deficiency but does not respond to iron therapy.
Recent Evidence Note
A 2024 systematic review (PMID 39267265) has characterized cuproptosis - a distinct copper-dependent cell death pathway involving copper accumulation in mitochondria, binding to lipoylated TCA cycle proteins, and proteotoxic stress - as a newly recognized mechanism with implications in cancer biology and neurodegeneration. This is an active area of research distinct from classical Wilson disease pathophysiology.
Sources: Sleisenger and Fordtran's Gastrointestinal and Liver Disease; Robbins & Kumar Basic Pathology; Harrison's Principles of Internal Medicine 22E; Medical Physiology (Boron & Boulpaep)
This is a shared conversation. Sign in to Orris to start your own chat.