Fabrys disease management
Reading File
Finding Sources
Searching PubMed
"Fabry disease" AND management
Searching the Web
Fabry disease management guidelines 2024 2025 enzyme replacement therapy
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Reading File
Reading File
Reading File
Reading File
Loading Image
Reading File
Reading File
Reading File
I have sufficient data to compose a comprehensive answer. Let me now write the full response.
Fabry Disease (Anderson-Fabry Disease) - Management
Background
Fabry disease is an X-linked lysosomal storage disorder caused by deficiency of alpha-galactosidase A (α-Gal A), encoded by the GLA gene on the X chromosome. This leads to progressive accumulation of globotriaosylceramide (Gb3) in vascular endothelial cells, podocytes, cardiomyocytes, and neurons throughout the body. Over 1,000 mutations have been described; classic disease involves <3% residual enzyme activity, while late-onset cardiac and renal variants retain >3% activity.
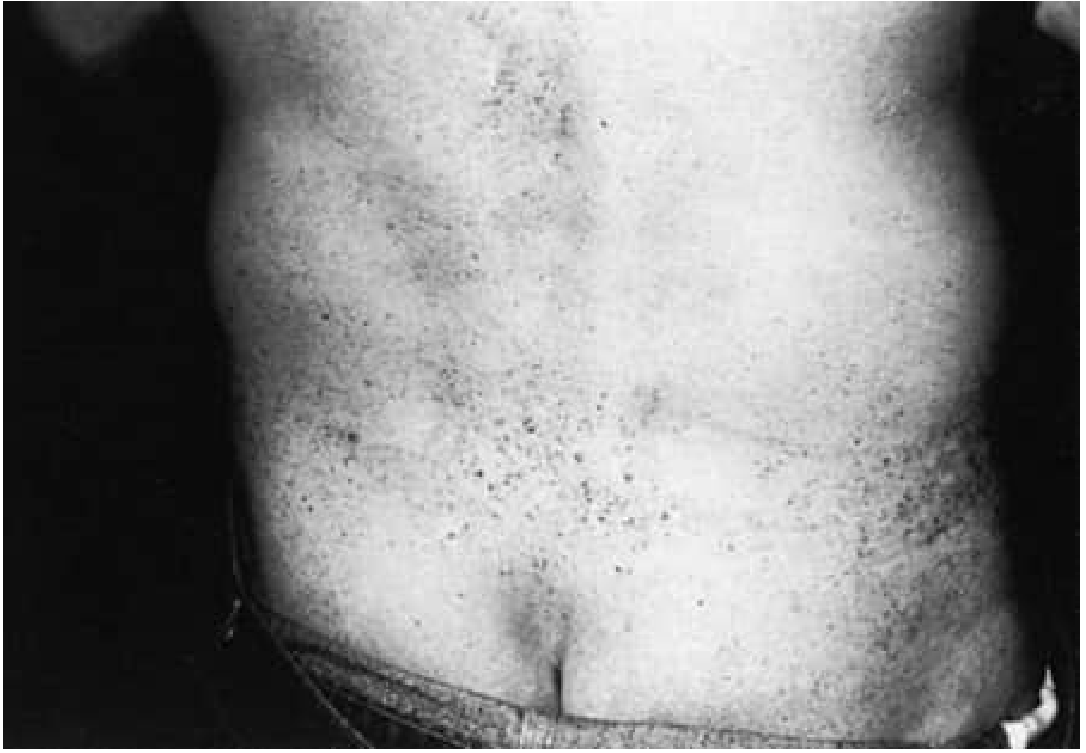
Disease-Specific Therapies
1. Enzyme Replacement Therapy (ERT)
ERT is the cornerstone of treatment. Two agents are established; a third (pegunigalsidase alfa) received approval more recently:
| Agent | Dose | Route | Notes |
|---|---|---|---|
| Agalsidase beta (Fabrazyme) | 1.0 mg/kg every 2 weeks | IV infusion | Produced in CHO cells; clears microvascular Gb3 deposits |
| Agalsidase alfa (Replagal) | 0.2 mg/kg every 2 weeks | IV infusion | Produced in human fibroblasts; improves neuropathic pain and cardiac/renal endpoints |
| Pegunigalsidase alfa (Elfabrio) | 1.0 mg/kg every 2 weeks | IV infusion | PEGylated enzyme; approved 2023; utilizes a different receptor mechanism than M6P pathway |
Key principles:
- ERT should be initiated before irreversible end-organ damage for maximum benefit - early treatment in men with classic phenotype yields the best renal and cardiac outcomes.
- Predicators of poorer renal response: high baseline proteinuria, worse baseline eGFR, delayed treatment initiation, history of cardiac or cerebrovascular events.
- ERT can be administered during dialysis sessions.
- Infusion reactions (flushing, chest discomfort, rash, pruritus, nasal congestion) are common; pre-medication and slowing infusion rate are standard management.
- Approximately 50-60% of male patients develop IgG antibodies to agalsidase beta; their long-term clinical significance is debated.
ERT initiation criteria (European Fabry Working Group / 2018 European recommendations):
- Classic males: treat at first symptoms or signs of organ involvement
- Females: treat when symptomatic (cardiac, renal, or neurological involvement)
- May be considered with eGFR <45 mL/min/1.73m², on dialysis, or with cognitive impairment
When to consider stopping ERT:
- Advanced end-organ damage with no further expected benefit
- Patient preference after informed discussion
- Severe infusion reactions not manageable with pre-medication
2. Oral Pharmacological Chaperone Therapy - Migalastat (Galafold)
Migalastat is an oral small molecule that reversibly binds the active site of α-Gal A, stabilizing misfolded protein and enabling correct lysosomal trafficking. It increases endogenous enzyme activity by 2-8 fold in amenable variants.
- Only for amenable GLA variants (specific missense mutations causing misfolding, confirmed via an in vitro cell-based assay - not all GLA variants are amenable)
- Dose: 123 mg orally every other day (to avoid competitive inhibition of the enzyme substrate)
- Age: Approved for adults ≥16 years
- Advantages: Oral, no infusion reactions, convenient, available in ambulatory settings
- The ATTRACT trial showed non-inferiority to ERT in stabilizing renal function in amenable-variant patients; also reduces LV mass index and GI symptoms
- Migalastat is not effective in patients with null mutations (frameshift, nonsense) or non-amenable missense variants
Supportive (Organ-Specific) Management
Renal
- ACE inhibitors or ARBs for proteinuria reduction (standard CKD management); note that recombinant α-Gal A may interact with endogenous ACE, causing transient BP drops during infusions
- Target BP <130/80 mmHg
- Kidney transplantation is preferred over dialysis for ESKD - Fabry nephropathy does not recur in the allograft, and transplant outcomes are better than dialysis. Kidney transplant is a first-choice therapy for Fabry-related kidney failure.
- Dialysis if transplant is not feasible
Cardiac
- LV hypertrophy monitoring: serial echocardiography and cardiac MRI (late gadolinium enhancement pattern in basal inferolateral wall is characteristic)
- Arrhythmia management: antiarrhythmics as indicated; pacemaker/ICD for heart block or malignant arrhythmia
- Hypertension: treat per standard guidelines; avoid beta-blockers in patients with existing sinus bradycardia
- Angina: manage small-vessel disease; epicardial coronary disease is uncommon
- ERT may stabilize or modestly regress LV hypertrophy when started early
Neurological - Neuropathic Pain
- Acute pain crises: NSAIDs, avoiding temperature extremes and triggering stressors
- Chronic neuropathic pain: gabapentin, carbamazepine, phenytoin, amitriptyline
- Stroke prevention: antiplatelet agents; aggressive vascular risk factor management (hypertension, dyslipidemia, smoking cessation, diabetes)
- TIAs and stroke most commonly involve the posterior circulation (vertebrobasilar)
Gastrointestinal
- Pain relief, H2-blockers or PPIs, prokinetic agents, pancreatic enzyme supplementation
- Dietary adjustments for postprandial bloating and early satiety
Skin (Angiokeratomas)
- Cosmetic removal with argon laser therapy (does not address underlying disease)
Hypohidrosis/Anhidrosis
- Avoidance of heat and exertion; cool environments; appropriate clothing
Psychiatric
- Depression is common: SSRIs or SNRIs are first-line
Dyslipidemia
- Routine surveillance and statin therapy per standard guidelines
Monitoring Parameters
Patients on ERT or migalastat require regular multidisciplinary follow-up:
| Parameter | Frequency |
|---|---|
| Urine protein/creatinine ratio | Every 6 months |
| eGFR | Every 6 months |
| Echocardiography / cardiac MRI | Annually |
| ECG (assess PR interval, arrhythmia) | Annually |
| Brain MRI | Every 2-3 years (or with new neurological symptoms) |
| Ophthalmology (slit-lamp, retinal vessels) | Annually |
| Audiometry | Annually |
| Lyso-Gb3 (plasma biomarker) | Every 6 months (treatment monitoring) |
| Urine Gb3 | Every 6 months |
Emerging and Investigational Therapies (as of 2025-2026)
- Substrate reduction therapy (SRT): inhibitors of glucosylceramide synthase (upstream precursor of Gb3) - reduce substrate synthesis rather than replacing or stabilizing enzyme
- mRNA therapy: periodic administration of mRNA encoding human α-Gal A (preclinical / early trials)
- AAV-based gene therapy: several trials actively recruiting; aims for durable α-Gal A expression
- Moss-derived α-galactosidase (moss-αGAL): plant-derived ERT with mannose-terminated glycans; shows strong kidney tissue affinity; early trials ongoing
- Second-generation enzyme formulations in development
Phenotypes and Treatment Timing
| Feature | Classic | Renal Variant | Cardiac Variant |
|---|---|---|---|
| Age at onset | 4-8 years | >25 years | >40 years |
| Enzyme activity | <1% | >1% | >1% |
| Angiokeratoma | ++ | - | - |
| Acroparesthesias | ++ | ± | - |
| Kidney | ESRD | ESRD | Proteinuria |
| Heart | LVH, ischemia | LVH | LVH, cardiomyopathy |
| Treatment | ERT early | ERT / migalastat | ERT / migalastat |
(Source: Braunwald's Heart Disease; NKF Primer on Kidney Diseases, 8e)
Diagnosis Triggers
Clinicians should suspect Fabry disease when two or more of the following are present in a young adult:
- Acroparesthesias/neuropathic pain since childhood
- Unexplained persistent proteinuria
- Hypertrophic cardiomyopathy with prominent diastolic dysfunction
- Progressive CKD of unknown cause
- Cryptogenic stroke or TIA
- X-linked family history of ESKD, stroke, or HCM
- Recurrent GI symptoms (nausea, diarrhea, abdominal pain)
Diagnosis in males: leukocyte α-Gal A activity (enzyme assay). In females: genetic testing is mandatory since enzyme activity may be normal due to X-inactivation.
Sources: Comprehensive Clinical Nephrology 7e, NKF Primer on Kidney Diseases 8e, Braunwald's Heart Disease 2 Vol Set, Bradley & Daroff's Neurology in Clinical Practice; BIMDG Fabry Clinical Guidance (June 2025); recent ERT/migalastat trial data (ATTRACT, Fabry Registry, FOS).
Recent evidence note (2025): A 2025 Orphanet J Rare Dis study (Lenders et al.) examined ERT vs. migalastat outcomes in female patients specifically, highlighting that female patients with amenable variants may achieve equivalent organ stabilization with oral migalastat. The 2025 BIMDG guidelines (June 2025) now include pegunigalsidase alfa (Elfabrio) as a first-line ERT option alongside agalsidase alfa and beta.
This is a shared conversation. Sign in to Orris to start your own chat.