Alzheimer' dieaes
Reading File
Finding Sources
Searching PubMed
"Alzheimer disease"[MeSH Terms] AND treatment
Reading File
Reading File
Reading File
Finding Sources
Reading File
Reading File
Reading File
Loading Image
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Finding Sources
Finding Sources
Searching the Web
lecanemab donanemab Alzheimer disease FDA approved 2024 2025 treatment update
I now have all the essential content to compile a thorough, well-cited answer. Let me synthesize everything.
Alzheimer's Disease - A Comprehensive Overview
1. Background and Definition
Alzheimer's disease (AD) was first described in 1906 by Alois Alzheimer, who identified the hallmark inclusions - amyloid plaques and neurofibrillary tangles (NFTs) - in a woman in her 50s with paranoia, memory loss, and aphasia. It is the most common cause of dementia, contributing to an estimated 60-70% of all dementia cases worldwide. Approximately 55 million people across the world currently live with dementia. In the United States alone, AD dementia care costs were estimated at $360 billion in 2024 (~$25,000 per patient).
The disease can manifest as early as the third decade of life, but is predominantly a disease of the elderly. Its definition has evolved: DSM-5 reclassifies it as "major neurocognitive disorder due to Alzheimer disease," and recent research frameworks now define AD biologically by the presence of both amyloid (A) and tau (T) biomarkers, irrespective of clinical symptoms.
- Harrison's Principles of Internal Medicine 22E, p. 3519
- Bradley and Daroff's Neurology in Clinical Practice
2. Epidemiology
| Age Group | Prevalence |
|---|---|
| 65-74 years | ~3% |
| 75-84 years | ~11% |
| 85+ years | ~32% |
-
Incidence rises sharply with age: 2 new cases/1,000 (age 65-74) to 37 new cases/1,000 (age 85+)
-
Lifetime risk from age 45: ~10% in men, ~20% in women
-
While MCI is more common in men, AD dementia prevalence is higher in women, partly explained by longer female life expectancy
-
Projections estimate ~15 million U.S. cases by 2060
-
Bradley and Daroff's Neurology in Clinical Practice
3. Pathology and Histology
The histopathological hallmarks are:
Neuritic Plaques
- Core of aggregated beta-amyloid (Aβ) peptide (39-42 amino acids) surrounded by degenerating neurites
- Aβ is proteolytically derived from the larger amyloid precursor protein (APP)
- Identified by decreased CSF Aβ42 or amyloid PET positivity
Neurofibrillary Tangles (NFTs)
- Intracellular aggregations of hyperphosphorylated tau protein (a microtubule-associated protein)
- Self-aggregate to form paired helical filaments
- Identified by tau PET or elevated phospho-tau in CSF
For a histopathological diagnosis, at least 6 neuritic plaques and neurofibrillary tangles per low-power field in frontal, temporal, or parietal cortex are required.
- Goldman-Cecil Medicine, p. 3863
4. Pathophysiology
The Amyloid Cascade
Beta-amyloidosis begins accumulating in the neocortex up to 20 years before dementia onset, detectable by PET imaging. Soluble oligomeric forms of Aβ are thought to be the key pathogenic molecules that induce neuronal injury. By the time clinical dementia appears, large numbers of neuritic plaques are present in the neocortex - but the amount of Aβ does not closely mirror dementia severity.
Tau Pathology and Spread
NFTs appear first in the medial temporal lobe and brainstem in cognitively normal persons as early as the fourth decade of life. The critical step in AD is transsynaptic spread of tangle pathology to cortical association areas:
- Mild symptoms → tangles concentrated in entorhinal cortex and hippocampus (episodic memory impairment)
- Moderate disease → tangles spread to frontal, parietal, and temporal association cortices
- Severe/late disease → occipital lobes and primary motor/sensory cortices
This spatial pattern of NFT spread closely parallels clinical symptom progression.
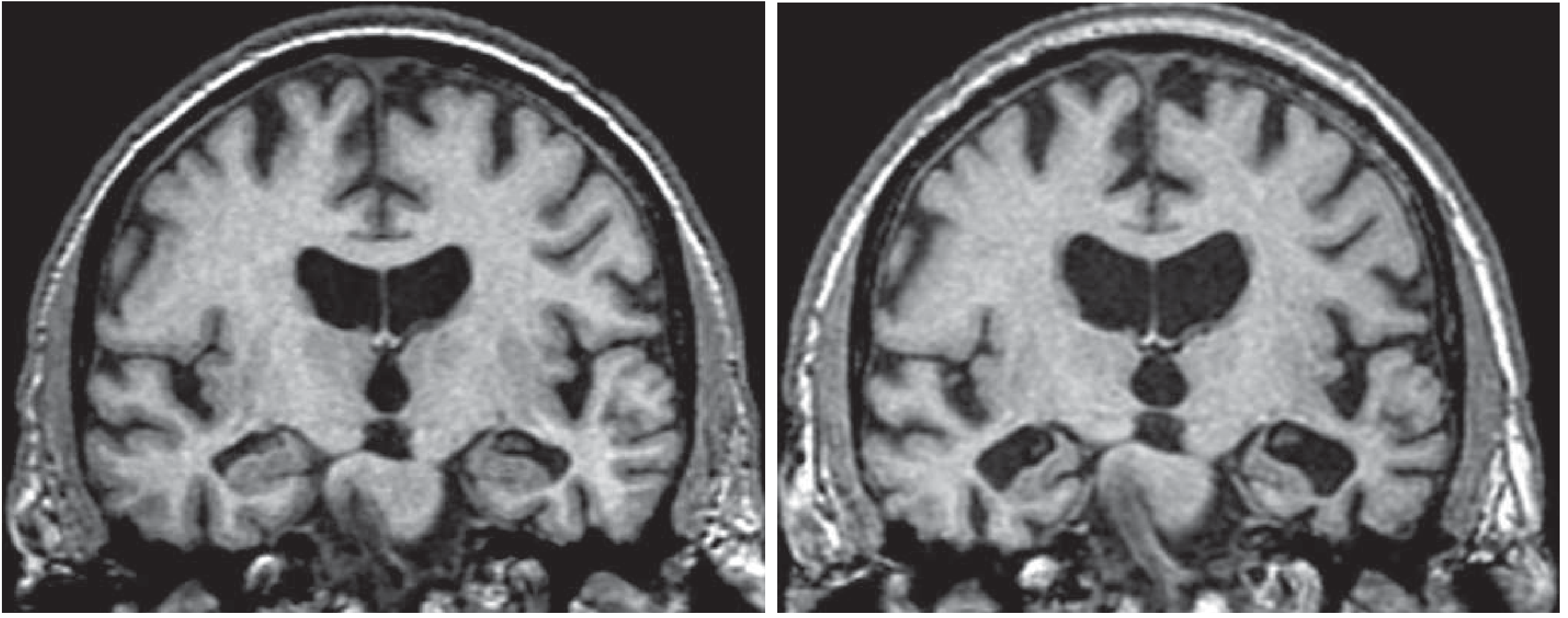
- Goldman-Cecil Medicine
5. Genetics
Autosomal Dominant (Early-Onset) - Rare (<1% of AD)
| Gene | Chromosome | Protein |
|---|---|---|
| APP | 21 | Amyloid precursor protein |
| PSEN1 | 14 | Presenilin-1 |
| PSEN2 | 1 | Presenilin-2 |
Sporadic (Late-Onset) Risk Genes
-
ApoE ε4 allele (chromosome 19): the single most important genetic risk factor for sporadic AD
- 1 ε4 allele: ~2-3x increased risk in women
- 2 ε4 alleles (homozygous): 10-15x increased risk in both sexes
- ~40-65% of AD patients carry at least one ε4 allele
- ApoE ε4 leads to less efficient amyloid clearance and may promote tau aggregation
- ApoE ε2 allele may be protective
-
TREM2 - inflammation-associated gene; heterozygous carriers have ~3x increased AD risk
-
Other GWAS-identified genes (CLU, PICALM, CR1) in pathways for innate immunity, lipid metabolism, and synaptic function
-
Harrison's Principles of Internal Medicine 22E
6. Clinical Manifestations
Typical Amnestic AD (most common)
- Insidious onset with episodic memory loss progressing to multi-domain cognitive decline
- Executive, language, and visuospatial deficits develop over years
- Anosognosia (impaired awareness of own deficits) is common
- Depression, social withdrawal, anxiety may precede cognitive symptoms
Atypical Presentations (~20% of AD)
- Posterior cortical atrophy syndrome (visual processing dysfunction)
- Logopenic aphasia (naming and repetition difficulties)
- Corticobasal syndrome (asymmetric akinetic-rigid-dystonic)
- Frontal/behavioral variant
Clinical Stages
| Stage | Description |
|---|---|
| Preclinical AD | Amyloid biomarker positivity, no symptoms; precedes symptoms by up to 20 years |
| Subjective cognitive decline | Self-perceived memory worsening, not yet measurable |
| MCI / Early symptomatic AD | Measurable objective decline, functionally compensated; ~12% per year progress to dementia |
| AD dementia | Cognitive impairment interfering with daily activities |
- Harrison's Principles of Internal Medicine 22E, pp. 3519-3523
7. Biomarker Framework (AT[N])
The current research framework classifies individuals by three biomarker groups:
| Biomarker | Positive Indicator | Method |
|---|---|---|
| A - Amyloid | ↓ CSF Aβ42 or amyloid PET positive | CSF, PET |
| T - Tau | Elevated CSF phospho-tau or tau PET positive | CSF, PET |
| N - Neurodegeneration | Hippocampal atrophy on MRI, ↓ FDG-PET | MRI, FDG-PET |
A+T+ = Alzheimer disease (regardless of clinical stage)
A+T- = Alzheimer pathological change only
- Bradley and Daroff's Neurology in Clinical Practice
8. Diagnosis
Clinical Diagnosis of Probable AD requires:
- Objective evidence of decline in memory AND at least one other cognitive domain
- Insidious onset and gradual progression
- No evidence of alternative etiology
- The definitive diagnosis requires postmortem neuropathological confirmation
Cognitive assessments: Neuropsychological testing (more sensitive than MRI for disease tracking). Classic findings include impaired recognition memory, intrusion errors, absence of primacy effect in word-list recall.
Neuroimaging:
- MRI: hippocampal atrophy (earliest reliable structural change)
- FDG-PET: hypometabolism in posterior cingulate, parietal, temporal cortices
- Amyloid PET and tau PET: detect pathological proteins in vivo
9. Treatment
Symptomatic Therapies
Cholinesterase Inhibitors (mild-moderate-severe AD)
These inhibit CNS acetylcholinesterase in the synaptic cleft, prolonging acetylcholine action. However, they do not halt neurodegeneration - benefit diminishes as presynaptic cholinergic neurons are progressively lost.
| Drug | Trade Name | Stage | Key Notes |
|---|---|---|---|
| Donepezil | Aricept | Mild-Moderate-Severe | Only ChEI approved for severe AD; noncompetitive reversible; t½=70h |
| Rivastigmine | Exelon | Mild-Moderate | Transdermal patch reduces GI side effects; dual AChE+BuChE inhibitor |
| Galantamine | Razadyne | Mild-Moderate | Also modulates nicotinic receptors; competitive reversible |
Memantine (moderate-severe AD)
-
NMDA receptor antagonist; reduces excitotoxic glutamate injury
-
Although commonly combined with a ChEI in clinical practice, trials have not consistently demonstrated cognitive efficacy in mild disease
-
Combined ChEI + memantine may provide better response than monotherapy
-
Kaplan & Sadock's Comprehensive Textbook of Psychiatry
Disease-Modifying Therapies (New - 2023-2025)
Anti-amyloid monoclonal antibodies represent a major advance:
- Lecanemab (Leqembi) - FDA approved 2023; targets soluble Aβ protofibrils; slows clinical decline by ~27% in early AD in phase III trials
- Donanemab (Kisunla) - FDA approved 2024; targets N-3pE-Aβ plaques; significant plaque clearance and slower decline in early AD
Key safety concern: ARIA (Amyloid-Related Imaging Abnormalities) - brain edema/microhemorrhages; risk is higher in ApoE ε4 carriers. ApoE genotyping is now recommended before starting these therapies.
10. Risk Factors and Prevention
Non-modifiable: Age (strongest), female sex, ApoE ε4 genotype, family history
Modifiable risk factors (Lancet Commission 2024): Hypertension, diabetes, obesity, smoking, physical inactivity, depression, low education, hearing loss, air pollution, social isolation, traumatic brain injury
Protective factors: Recent meta-analysis (PMID 39797935) found the Mediterranean diet significantly associated with reduced risk of cognitive impairment and AD. Physical exercise, cognitive engagement, and management of cardiovascular risk factors are also consistently supported.
11. Recent Evidence (2024-2026)
- Updated amyloid and tau PET appropriate use criteria were released by the Alzheimer's Association and SNMMI in 2025 (PMID 39776249)
- LATE (Limbic-predominant Age-related TDP-43 Encephalopathy) clinical criteria were published in 2025 (PMID 39807681) - an important AD mimic that co-occurs with AD and should be considered in older patients
- Non-invasive brain stimulation + cognitive training shows promise as adjunct therapy in MCI/AD (meta-analysis, PMID 38937842)
Key References:
- Harrison's Principles of Internal Medicine, 22nd Ed. (2025), Chapter on Alzheimer's Disease
- Goldman-Cecil Medicine, International Edition (2-vol set), Chapter 371
- Bradley and Daroff's Neurology in Clinical Practice, Chapters 44 & 95
- Kaplan & Sadock's Comprehensive Textbook of Psychiatry, Chapter 55
This is a shared conversation. Sign in to Orris to start your own chat.