Factors modifying drug actions
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Finding Sources
Reading File
Finding Sources
I now have enough material from the library. Let me compile a thorough answer.
Factors Modifying Drug Actions
Drug action is not uniform across all individuals or all circumstances. Many patient-specific, physiological, genetic, and environmental factors can alter a drug's pharmacokinetics (what the body does to the drug) or pharmacodynamics (what the drug does to the body), resulting in variable responses - from therapeutic success to toxicity or complete failure.
1. Pharmacogenetic and Genetic Factors
Individual drug response depends on the complex interplay of drug factors (dose, route, formulation), environmental factors (diet, infections, other drugs, exercise, toxins), clinical factors (age, organ function), and genetic factors.
Key genes involved in driving variable drug actions are called pharmacogenes. They influence drug response via:
- Pharmacokinetics - variable drug concentrations
- Pharmacodynamics - variable drug effects
Pharmacogenes encode:
- Drug-metabolizing enzymes (e.g., CYP2D6, CYP2C19, CYP2C9)
- Drug transport molecules (e.g., P-glycoprotein, SLCO1B1)
- Drug targets (e.g., beta-adrenoceptor variants)
- Genes modulating the molecular context of disease
Metabolizer Phenotypes
Drug metabolism traits can follow a "monogenic" pattern, producing distinct groups:
| Phenotype | Genotype | Clinical Effect |
|---|---|---|
| Poor metabolizer | Two non-functional alleles | Drug accumulates → toxicity risk |
| Intermediate metabolizer | One functional + one non-functional allele | Reduced clearance |
| Normal (extensive) metabolizer | Two functional alleles | Standard response |
| Ultra-rapid metabolizer | Gene duplication | Rapid clearance → therapeutic failure |
Classic Examples
- G6PD deficiency - Hemolysis with antimalarials (primaquine), sulfonamides (pharmacodynamic)
- Pseudocholinesterase deficiency - Prolonged paralysis after succinylcholine
- Malignant hyperthermia - Ryanodine receptor (RYR1) mutation with volatile anaesthetics
- NAT (N-acetyltransferase) polymorphism - Slow vs. fast acetylators affect INH, hydralazine, procainamide
- CYP2D6 variants - Affect codeine, metoprolol, tricyclic antidepressants, tamoxifen
Source: Goodman & Gilman's The Pharmacological Basis of Therapeutics
2. Age
Age profoundly affects both pharmacokinetics and pharmacodynamics.
Neonates and Infants
- Absorption: Gastric pH is higher (less acid), affecting absorption of weakly acidic/basic drugs; slower gastric emptying
- Distribution: Higher body water, lower plasma protein (albumin), higher free drug fractions; immature blood-brain barrier
- Metabolism: Hepatic CYP enzymes immature at birth (especially CYP3A4, glucuronidation); chloramphenicol "Grey Baby Syndrome" - underdeveloped conjugation enzymes lead to toxic accumulation
- Excretion: Glomerular filtration rate (GFR) is ~30% of adult value at birth; full maturation by 6-12 months
Elderly
- Absorption: Reduced gut motility; reduced first-pass metabolism
- Distribution: Reduced lean body mass, increased fat (increased Vd for lipid-soluble drugs); reduced albumin (higher free drug)
- Metabolism: Hepatic blood flow and enzyme activity decline by ~30-40%
- Excretion: GFR declines with age (~1 mL/min per year after 40); serum creatinine may not reflect this due to reduced muscle mass
- Pharmacodynamics: CNS more sensitive to sedatives, opioids, anticoagulants; polypharmacy increases interaction risk
3. Body Weight and Body Composition
- Doses for obese patients: fat-soluble drugs (e.g., diazepam) have a larger volume of distribution
- Water-soluble drugs (e.g., aminoglycosides) are dosed on lean body weight, not total body weight
- Doses in pediatrics are typically calculated in mg/kg or by body surface area (mg/m²) for chemotherapy
4. Sex (Biological)
- Women generally have lower body weight, less lean mass, lower CYP3A4 activity (though CYP3A4 is actually slightly higher in women)
- CYP2D6 and CYP2C19 show some sex-based differences
- Women are more sensitive to some CNS drugs (anesthetics, analgesics)
- QT prolongation risk from antiarrhythmics (e.g., sotalol, quinidine) is greater in women due to longer baseline QT
- Hormonal changes in pregnancy significantly alter pharmacokinetics (see below)
5. Pregnancy
- Absorption: Delayed gastric emptying, nausea/vomiting alter oral bioavailability
- Distribution: Plasma volume expands by ~50%, altering Vd; albumin falls (more free drug); additional compartment (fetal-placental)
- Metabolism: CYP3A4, CYP2C9 activity increases; CYP1A2 and CYP2C19 decrease
- Excretion: GFR increases by ~50%; renal drug clearance increases (e.g., ampicillin, digoxin need dose adjustment)
- Fetal risk: Teratogens act on the fetus via placental transfer (e.g., thalidomide, phenytoin, warfarin)
6. Disease States
Renal Disease
- Reduced clearance of renally excreted drugs and active metabolites
- Dose reduction or interval extension required (creatinine clearance guides adjustment)
- Examples: aminoglycosides, digoxin, NSAIDs (risk of acute kidney injury), lithium, metformin (contraindicated in severe CKD)
Hepatic Disease
- Reduced first-pass metabolism → increased bioavailability of oral drugs (e.g., propranolol, morphine)
- Reduced synthesis of plasma proteins (lower albumin, higher free drug fraction)
- Portosystemic shunts bypass hepatic metabolism entirely
- Reduced bile production affects absorption of fat-soluble drugs and enterohepatic circulation
- Examples requiring caution: benzodiazepines, opioids, paracetamol, warfarin
Gastrointestinal Disease
- Malabsorption (Crohn's disease, coeliac disease) reduces drug absorption
- Altered gut motility (diarrhoea or constipation) affects absorption rate
Cardiovascular Disease
- Reduced cardiac output → reduced hepatic and renal blood flow → reduced drug clearance
- Edematous states alter Vd of water-soluble drugs
Thyroid Disease
- Hypothyroidism slows metabolism; hyperthyroidism increases it
- Hypothyroidism increases sensitivity to digoxin; hyperthyroidism decreases it
7. Drug Interactions
Drugs can modify each other's actions at multiple levels:
Pharmacokinetic Interactions
- Absorption: Antacids reduce absorption of tetracyclines, fluoroquinolones (chelation); metoclopramide increases gastric emptying (faster absorption)
- Protein binding displacement: Warfarin displaced by NSAIDs - transient increase in free warfarin
- Enzyme induction: Rifampicin, carbamazepine, phenytoin induce CYP enzymes → reduced levels of co-administered drugs (OCP failure, reduced anticoagulation)
- Enzyme inhibition: Erythromycin, ketoconazole, grapefruit juice inhibit CYP3A4 → increased drug levels and toxicity (e.g., statins → myopathy; ciclosporin toxicity)
- Transporter interactions: P-gp inhibitors (e.g., amiodarone) raise digoxin levels
Pharmacodynamic Interactions
- Synergism/Additive: Alcohol + benzodiazepines (CNS depression); aspirin + heparin (additive bleeding)
- Antagonism: Beta-blockers antagonize salbutamol; naloxone reverses opioids
- Receptor level: Warfarin and vitamin K compete at clotting factor synthesis
8. Route and Formulation of Administration
- Oral: Subject to first-pass metabolism; slower onset; affected by food, gastric pH, motility
- Intravenous: 100% bioavailability; immediate onset; no first-pass effect
- Sublingual: Bypasses first-pass (e.g., glyceryl trinitrate, buprenorphine)
- Transdermal: Slow, sustained delivery; bypasses GI and first-pass (e.g., fentanyl patch, nicotine patch)
- Inhaled: Rapid onset for pulmonary/systemic effects; local action in airways (e.g., salbutamol)
- Modified release formulations (SR/XR/CR): Alter release kinetics, reduce dosing frequency, smooth concentration-time profiles
Food interactions:
- High-fat meals increase absorption of lipophilic drugs (e.g., griseofulvin, itraconazole)
- Grapefruit juice irreversibly inhibits intestinal CYP3A4
- Tyramine-rich foods + MAOIs → hypertensive crisis
9. Tolerance, Tachyphylaxis, and Dependence
| Term | Definition | Example |
|---|---|---|
| Tolerance | Reduced response to a drug on repeated administration; requires increasing dose for same effect | Opioids, nitrates, benzodiazepines |
| Tachyphylaxis | Rapid tolerance developing after just a few doses (acute tolerance) | Ephedrine, LSD, amphetamines |
| Cross-tolerance | Tolerance to one drug confers tolerance to pharmacologically related drugs | Opioids, benzodiazepines |
| Dependence | Physical/psychological need for drug; withdrawal syndrome on cessation | Opioids, alcohol, benzodiazepines |
Mechanisms of tolerance:
- Receptor downregulation (decreased receptor density)
- Receptor desensitization (uncoupling of receptor from effector)
- Increased metabolic degradation (pharmacokinetic tolerance)
- Compensatory physiological mechanisms (counterregulation)
10. Psychological Factors - Placebo Effect
- A placebo (inert substance) can produce measurable physiological effects through expectation and conditioning
- The placebo effect can account for 30-40% of the analgesic response in clinical trials
- Nocebo effect: Negative expectations worsen outcomes or produce side effects
- These factors are accounted for in double-blind, randomized controlled trials
11. Cumulation and Dose-Related Factors
- Drugs with long half-lives or narrow therapeutic indices accumulate with repeated dosing if elimination is impaired
- Steady-state concentration is reached in ~5 half-lives; influenced by dose, frequency, and clearance
- Therapeutic drug monitoring (TDM) is used for drugs with narrow therapeutic windows (e.g., digoxin, phenytoin, lithium, aminoglycosides, vancomycin)
12. Environmental and Lifestyle Factors
| Factor | Effect |
|---|---|
| Smoking | Induces CYP1A2 → reduces levels of theophylline, clozapine, olanzapine |
| Alcohol (chronic use) | Induces CYP2E1 → increased paracetamol hepatotoxicity |
| Alcohol (acute use) | Inhibits CYP enzymes → increases drug levels |
| Diet | High-protein diet induces drug metabolism; grapefruit CYP3A4 inhibition |
| Exercise | Alters blood flow, absorption, and distribution |
| Altitude | Alters drug absorption (hypoxia-induced GI changes) |
Summary Table: Key Factors at a Glance
| Factor | Primary Pharmacokinetic Impact | Pharmacodynamic Impact |
|---|---|---|
| Age (pediatric/elderly) | Altered metabolism & excretion | Altered receptor sensitivity |
| Renal disease | Reduced excretion | - |
| Hepatic disease | Reduced metabolism, protein binding | - |
| Genetics (CYP polymorphism) | Altered metabolism | Altered receptor variants |
| Pregnancy | Increased Vd, altered metabolism | Teratogenicity |
| Drug interactions | ADME alterations | Synergism/antagonism |
| Route of administration | Bioavailability, onset | - |
| Tolerance | - | Receptor downregulation |
| Body weight/composition | Vd alteration | - |
| Smoking/diet | Enzyme induction/inhibition | - |
Understanding these modifying factors is fundamental to individualized (precision) pharmacotherapy - selecting the right drug, at the right dose, via the right route, for the right patient.
Sources: Goodman & Gilman's The Pharmacological Basis of Therapeutics; Katzung's Basic and Clinical Pharmacology, 16th Edition; Goldman-Cecil Medicine
Routes of drug administration
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
I have comprehensive content from both Lippincott and Goodman & Gilman, plus an excellent diagram. Here is the full answer:
Routes of Drug Administration
The route of administration is determined by the drug's physicochemical properties (water/lipid solubility, ionization, molecular size, stability) and the therapeutic objective (speed of onset, duration, local vs systemic effect, patient condition).
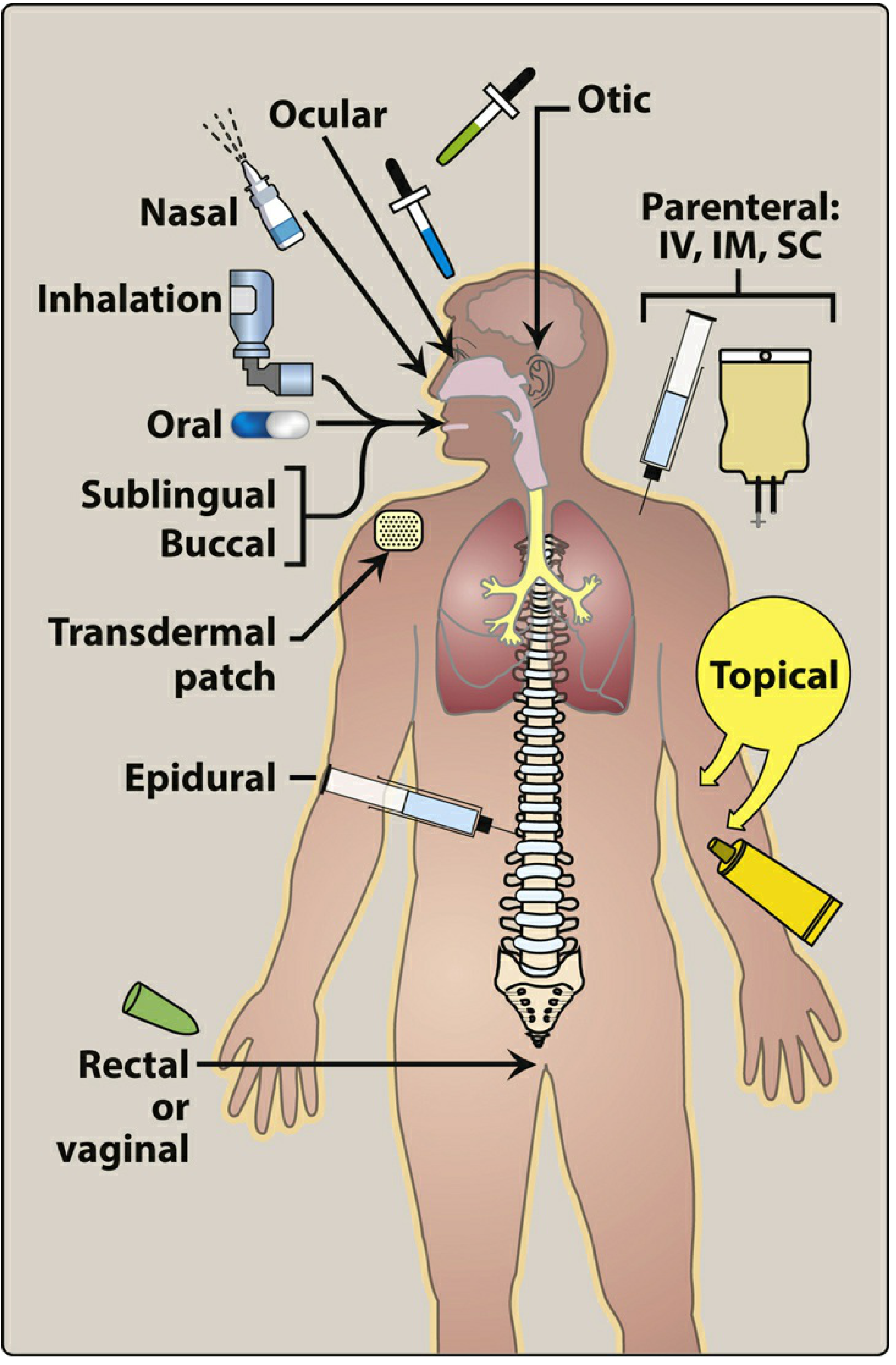
Figure: Commonly used routes of drug administration - Lippincott Illustrated Reviews: Pharmacology
Routes are broadly classified into three categories: Enteral, Parenteral, and Other (Topical/Special).
I. ENTERAL ROUTES
Enteral administration involves the GI tract. It is the most common, convenient, and economical method.
1. Oral (Per Oral / PO)
Mechanism of absorption: Primarily passive diffusion through the GI mucosa. Most absorption occurs in the small intestine due to its large surface area and rich blood supply. Nonionized, lipophilic drugs are better absorbed.
Bioavailability (F): Variable - 0.05 < F < 1, highly dependent on drug properties and GI factors.
Advantages:
- Safest, most convenient, most economical
- Self-administration possible
- Large variety of formulations available
- Overdose can be managed (e.g., activated charcoal, gastric lavage)
Disadvantages:
- Irregular and incomplete absorption for some drugs
- Inactivation by gastric acid (e.g., penicillin G, insulin)
- Destruction by digestive enzymes
- First-pass metabolism by intestinal mucosa, gut microbiome, and liver - significantly reduces bioavailability
- Requires patient cooperation/compliance
- Vomiting and food can alter absorption
- Unsuitable for unconscious or vomiting patients
Factors affecting GI absorption:
- Surface area and blood flow at absorption site
- Drug solubility (rate-limiting step for solid forms is dissolution)
- Gastric pH (weak acids absorbed better in stomach pH 1-2; weak bases absorbed better in intestine pH 5-7)
- Gastric emptying rate: faster emptying generally increases absorption rate
- Presence of food (can delay, reduce, or increase absorption)
- Gut microbiome (>1000 species can metabolize drugs)
Special oral formulations:
| Formulation | Mechanism | Purpose | Example |
|---|---|---|---|
| Enteric-coated | Chemical coating dissolves in alkaline intestine, not acid stomach | Protect acid-labile drugs; avoid gastric irritation | Omeprazole, aspirin EC |
| Extended-release (ER/XR/SR/CR) | Special coatings/matrix control drug release rate | Slower absorption, prolonged action, fewer doses/day, smoother plasma levels, better compliance | Morphine SR, metoprolol XL, nifedipine CR |
Controlled-release preparations are most suitable for drugs with short half-lives (t½ < 4 h). - Goodman & Gilman's
2. Sublingual
Drug is placed under the tongue. Absorbed directly into systemic venous circulation through the highly vascular sublingual mucosa.
Bioavailability: High for suitable drugs (e.g., nitroglycerin ~100%)
Advantages:
- Rapid onset of action (onset within 1-2 minutes for nitroglycerin)
- Completely bypasses first-pass hepatic metabolism
- Avoids destruction by gastric acid and digestive enzymes
- Saliva pH is ~7 (neutral) - drug stability maintained
Disadvantages:
- Limited to small doses
- Only suitable for drugs with high lipid solubility and low molecular weight
- Some drug is lost by swallowing saliva
Examples: Nitroglycerin (angina), buprenorphine, fentanyl (pain), ergotamine
3. Buccal
Drug is placed between the cheek and gum. Similar advantages to sublingual - bypasses first-pass metabolism and gastric acid.
Examples: Buccal midazolam (for seizures), testosterone buccal tablets
4. Rectal
Drug is administered via suppository or enema into the rectum.
Key pharmacokinetic point: Approximately 50% of the rectal venous drainage bypasses the portal circulation (via inferior and middle rectal veins → internal iliac → inferior vena cava), reducing but not eliminating first-pass metabolism.
Advantages:
- Useful when patient is vomiting, unconscious, or unable to swallow
- Avoids GI enzyme destruction
- Partial bypass of first-pass effect
- Useful for local anorectal conditions
Disadvantages:
- Absorption is erratic and incomplete
- Many drugs irritate the rectal mucosa
- Psychologically unacceptable to some patients
Examples: Diazepam (status epilepticus in children), paracetamol, mesalamine (ulcerative colitis), ondansetron
II. PARENTERAL ROUTES
Parenteral means "outside the alimentary canal." Drugs are introduced directly into tissues or the bloodstream, bypassing the GI tract entirely.
General indications for parenteral use:
- Drugs poorly absorbed or unstable in the GI tract (heparin, insulin, aminoglycosides)
- Unconscious or uncooperative patients
- Need for rapid onset (emergencies)
- Need for precise dose control
General disadvantages:
- Irreversible once administered
- Requires sterile technique (risk of infection)
- Pain, fear, local tissue damage
- Trained personnel usually required (except SC/IM self-injection)
- Cannot be recalled or neutralized like oral overdose
1. Intravenous (IV)
Drug is injected directly into a vein.
Bioavailability: F = 1.0 by definition (100% - absorption is circumvented).
Modes:
- IV bolus: Entire dose delivered rapidly; highest immediate plasma concentration; used in emergencies
- IV infusion: Drug infused over time; lower peak concentrations; more controlled; used for drugs requiring steady plasma levels (e.g., heparin, vancomycin, chemotherapy)
Advantages:
- Fastest onset of action (seconds)
- Maximum control over plasma levels; allows precise dose titration
- Suitable for large volumes
- Suitable for irritating drugs (diluted in large volume)
- Suitable for high-molecular-weight proteins/peptides (e.g., monoclonal antibodies)
- Essential in emergencies
Disadvantages:
- No recall possible - adverse reactions immediate and severe
- Risk of thrombophlebitis, embolism, infection (septicaemia)
- Must inject slowly as a rule (to avoid cardiovascular collapse)
- Oily solutions or poorly water-soluble drugs cannot be given IV
- Strict aseptic technique required
Examples: Morphine (acute pain/MI), furosemide (pulmonary oedema), adrenaline (anaphylaxis), most chemotherapy agents
2. Intramuscular (IM)
Drug is injected into skeletal muscle (deltoid, gluteus maximus, vastus lateralis).
Bioavailability: 0.75 < F < 1
Absorption: Via simple diffusion into capillaries; rate depends on:
- Drug solubility: aqueous solutions absorbed rapidly; oily/depot preparations absorbed slowly and sustainedly
- Blood flow to the muscle (reduced in shock)
Depot preparations: Drug suspended in non-aqueous vehicle (oil, polyethylene glycol). Vehicle diffuses out, drug precipitates at site, then dissolves slowly - provides sustained drug release over days to weeks.
Advantages:
- Moderate volumes can be given (up to 5 mL)
- Suitable for oily vehicles and some irritating substances
- Depot formulations provide prolonged effect (useful for antipsychotics, contraceptives)
- Suitable for self-injection (e.g., insulin, some vaccines)
Disadvantages:
- Possible pain or necrosis with irritating drugs
- Contraindicated during anticoagulant therapy (risk of haematoma)
- Absorption unreliable in shock (reduced blood flow)
- May falsely elevate creatine kinase (CK) on blood tests
- Accidental intravenous injection possible
Examples: Depot antipsychotics (fluphenazine decanoate, haloperidol decanoate), depo-medroxyprogesterone (contraception), IM vaccines, penicillin G benzathine
3. Subcutaneous (SC / SQ)
Drug is injected into the loose connective tissue beneath the skin.
Bioavailability: 0.75 < F < 1
Absorption: Via simple diffusion; slower than IM due to less vascularity; can be further slowed by adding vasoconstrictors (e.g., adrenaline with local anaesthetics).
Advantages:
- Slow, constant, sustained absorption
- Less risk of haemolysis or thrombosis compared to IV
- Suitable for self-injection
- Can be used for implants (e.g., hormonal implants, insulin pumps)
Disadvantages:
- Small volumes only (~2 mL maximum)
- Not suitable for irritating or hypertonic solutions (severe pain and tissue necrosis)
- Absorption unreliable in poor peripheral perfusion
Examples: Insulin, heparin (LMWH), adrenaline (anaphylaxis kit), some vaccines, hormonal implants (etonogestrel)
4. Intradermal (ID)
Drug is injected into the dermis (more vascular layer beneath the epidermis), producing a characteristic bleb.
Volume: Very small (0.1 mL)
Uses:
- Allergy skin testing (tuberculin/Mantoux test, allergen testing)
- Diagnostic skin tests
- Desensitisation (immunotherapy)
- BCG vaccine
Examples: Tuberculin (PPD), BCG, allergen testing solutions
5. Intra-arterial
Drug is injected directly into an artery to deliver high concentrations to a specific organ before systemic dilution.
Uses:
- Intra-arterial chemotherapy (e.g., hepatic artery infusion for liver metastases)
- Contrast dye injection for angiography
- Thrombolytic therapy (e.g., intra-arterial tPA for stroke)
Risk: Serious - can cause arterial spasm, thrombosis, ischaemia distally
6. Intrathecal / Intraventricular
Drug is injected into the subarachnoid space (intrathecal) or directly into a cerebral ventricle (intraventricular).
Rationale: The blood-brain barrier (BBB) prevents many drugs from reaching the CNS. Intrathecal delivery bypasses the BBB entirely.
Uses:
- Spinal anaesthesia (bupivacaine, lidocaine)
- Intrathecal chemotherapy (methotrexate for CNS lymphoma/leukaemia)
- Antibiotics for meningitis when systemic levels are insufficient
- Intrathecal baclofen (severe spasticity)
- Opioids for intractable pain
7. Intraosseous (IO)
Drug or fluid is injected directly into the bone marrow cavity (usually tibial plateau or sternum).
Use: Emergency vascular access when IV access is impossible (e.g., paediatric resuscitation, cardiac arrest). Bone marrow communicates with central circulation rapidly.
8. Epidural / Intradural
Drug is injected into the epidural space (between the ligamentum flavum and dura mater).
Uses:
- Epidural anaesthesia/analgesia in labour and surgery
- Epidural corticosteroid injections for disc prolapse/radicular pain
- Epidural opioids for postoperative/cancer pain
III. TOPICAL AND OTHER ROUTES
1. Topical
Drug is applied to skin or mucous membranes for local action at the site of application, with minimal systemic absorption desired.
Examples: Corticosteroid creams (eczema), antifungal creams, eye drops, ear drops, nasal sprays (for local rhinitis), mouth washes
2. Transdermal
Drug is applied to skin (usually via a patch) for systemic absorption.
Mechanism: Drug diffuses through skin layers into dermal capillaries. Rate of absorption depends on:
- Lipid solubility of the drug (must penetrate the stratum corneum)
- Skin thickness and integrity at the application site
- Surface area of contact
- Drug concentration in the vehicle
Advantages:
- Avoids first-pass metabolism
- Prolonged, steady plasma levels (zero-order kinetics)
- Non-invasive, convenient, improved compliance
- Easily removable in case of adverse reaction
Disadvantages:
- Only suitable for potent, lipid-soluble drugs
- Slow onset (hours to reach therapeutic levels)
- Local skin irritation or allergic contact dermatitis
- Cannot deliver large doses
Examples:
| Drug | Indication | Patch Duration |
|---|---|---|
| Glyceryl trinitrate (GTN) | Angina prophylaxis | 24 hours |
| Fentanyl | Chronic cancer/non-cancer pain | 72 hours |
| Nicotine | Smoking cessation | 16-24 hours |
| Estradiol | HRT, contraception | Variable |
| Buprenorphine | Pain | 7 days |
| Clonidine | Hypertension | 7 days |
| Scopolamine | Motion sickness | 3 days |
3. Inhalation
Drug is inhaled as a gas, vapour, or aerosol to act locally in the lungs or for rapid systemic absorption.
Why rapid? The pulmonary epithelium has an enormous surface area (~70 m²) and an extremely rich blood supply; drug crosses a very thin membrane to reach the circulation - onset nearly as fast as IV bolus.
Forms:
- Pressurised metered-dose inhalers (pMDI)
- Dry powder inhalers (DPI)
- Nebulisers
- Volatile anaesthetic gases
Advantages:
- Rapid onset
- Local delivery to the lung - high local concentrations with low systemic side effects (e.g., inhaled corticosteroids)
- Effective for gases (volatile anaesthetics, oxygen)
Disadvantages:
- Requires patient coordination and technique (pMDI)
- Dose difficult to calculate precisely
- Aerosol deposition unpredictable
- Local adverse effects (candidiasis with inhaled steroids)
Examples: Salbutamol (asthma), beclometasone (inhaled steroid), tiotropium (COPD), volatile anaesthetics (isoflurane, sevoflurane)
4. Nasal
Drug is administered directly into the nasal cavity.
Uses:
- Local: allergic rhinitis (fluticasone nasal spray), nasal decongestants (xylometazoline)
- Systemic: desmopressin (diabetes insipidus), calcitonin, sumatriptan, midazolam (for seizures - intranasal), naloxone (overdose reversal)
Advantage: Avoids first-pass metabolism; rapid onset for systemic drugs (highly vascular nasal mucosa).
5. Ocular
Drug is instilled as eye drops or ointment into the conjunctival sac.
Primarily for local effect (glaucoma drops, antibiotics for conjunctivitis) but systemic absorption can occur via the nasolacrimal duct (clinical significance: beta-blocker eye drops causing bradycardia/bronchospasm).
6. Vaginal
Drugs administered as pessaries, creams, or rings for local or systemic effect.
Examples: Clotrimazole (local antifungal), misoprostol (cervical ripening), progesterone gel (IVF support), combined hormonal vaginal ring (NuvaRing - contraception, systemic)
IV. COMPARISON SUMMARY TABLE
| Route | Bioavailability | Onset | First-Pass | Key Use |
|---|---|---|---|---|
| IV | 100% (F = 1) | Seconds | None | Emergencies, precise control |
| IM | 75-100% | Minutes | None | Depot drugs, vaccines |
| SC | 75-100% | Minutes-hours | None | Insulin, heparin, implants |
| Intradermal | Low (intentional) | - | None | Allergy testing, vaccines |
| Oral | Variable (5-100%) | 30-90 min | Yes | Routine outpatient therapy |
| Sublingual | High for lipophilic | 1-5 min | None | Nitroglycerin, buprenorphine |
| Rectal | Partial (~50% bypasses) | 15-30 min | Partial | Vomiting, unconscious patients |
| Transdermal | Variable | Hours | None | Prolonged systemic effect |
| Inhalation | Variable | Seconds-minutes | None | Respiratory disease, anaesthesia |
| Intrathecal | N/A (direct CNS) | Minutes | None | Spinal anaesthesia, CNS infections |
| Nasal | Good for small molecules | Minutes | None | Systemic/local nasal drugs |
V. FIRST-PASS METABOLISM - KEY CONCEPT
The first-pass effect (presystemic elimination) is the metabolism of a drug in the gut wall and liver before it reaches systemic circulation. It is relevant to all routes where blood drains into the portal circulation (oral, rectal to a degree).
Routes that AVOID first-pass metabolism:
IV > Inhalation > Sublingual/Buccal > Transdermal > Nasal > Rectal (partial) > IM/SC
Clinical implication: Drugs with high first-pass extraction (e.g., morphine, propranolol, GTN, lignocaine) need much smaller parenteral doses than oral doses. For example, oral morphine bioavailability is ~30% but IV is 100%.
Sources: Lippincott Illustrated Reviews: Pharmacology; Goodman & Gilman's The Pharmacological Basis of Therapeutics
Plasma half life shortly
Finding Sources
Reading File
I have the content needed. Here is a concise, clear answer:
Plasma Half-Life (t½)
Definition
The plasma half-life (t½) is the time required for the plasma concentration of a drug to fall to 50% of its current value.
For a first-order process:
t½ = 0.693 × Vd / CL
Where:
- Vd = Volume of distribution
- CL = Clearance
- 0.693 = ln 2 (natural log of 2)
Key Properties
- Most drug elimination follows first-order kinetics - a constant fraction (not amount) is eliminated per unit time
- Half-life is constant regardless of drug concentration (in first-order kinetics)
- After 4-5 half-lives, a process (elimination or accumulation) is considered essentially complete (~97%)
Drug Elimination Over Time
| Half-lives elapsed | % Drug eliminated | % Remaining |
|---|---|---|
| 1 | 50% | 50% |
| 2 | 75% | 25% |
| 3 | 87.5% | 12.5% |
| 4 | 93.75% | 6.25% |
| 5 | 96.9% | ~3% |
Clinical Significance
- Dosing interval: Drugs with short t½ need more frequent dosing; long t½ allows once-daily dosing
- Time to steady state: Steady-state plasma concentration is reached after ~5 half-lives, regardless of dose or dosing frequency
- Duration of action: A longer t½ generally means a longer duration of effect
- Drug washout: After stopping a drug, it is effectively eliminated in ~5 half-lives (important before switching drugs, e.g., MAOIs)
- Loading dose: Used when rapid steady state is needed for drugs with long t½ (e.g., amiodarone, digoxin)
Zero-Order Kinetics (Exception)
A few drugs (e.g., phenytoin, ethanol, aspirin at high doses) follow zero-order kinetics - a fixed amount is eliminated per unit time (enzyme saturation). In this case:
- t½ is not constant - it increases as concentration rises
- Small dose increases can cause disproportionately large rises in plasma levels → toxicity risk
Examples of Drug Half-Lives
| Drug | t½ | Clinical implication |
|---|---|---|
| Aspirin | 15-20 min | Metabolite (salicylate) t½ is much longer |
| Penicillin G | ~30 min | Requires frequent IV dosing |
| Gentamicin | 2-3 h | Once-daily dosing used clinically |
| Digoxin | 36-48 h | Takes ~7-10 days to reach steady state |
| Amiodarone | Weeks-months | Very prolonged effect; slow washout |
| Fluoxetine | 1-4 days | No washout period needed before most drugs |
Summary: t½ governs dosing frequency, time to steady state, and duration of drug effect. It is determined by both the volume of distribution and clearance - any disease or factor that increases Vd or reduces CL will prolong t½.
Source: Harrison's Principles of Internal Medicine, 22nd Edition
Drug tolerance
Finding Sources
Finding Sources
Finding Sources
Reading File
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Excellent content. Here is the comprehensive answer:
Drug Tolerance
Definition
Drug tolerance is a state in which repeated administration of the same dose produces a diminishing effect, or conversely, increasing doses are required to produce the same effect. It represents a rightward shift of the dose-response curve.
"Tolerance is a rightward shift of the dose-response curve and is a state in which an increased dosage of a psychoactive substance is needed to produce a desired effect." - Barash's Clinical Anesthesia
Types of Tolerance
1. Innate (Natural) Tolerance
- Genetically predetermined, pre-existing reduced sensitivity to a drug
- Present before any drug exposure
- Example: Certain individuals have naturally low sensitivity to alcohol due to genetic variants in alcohol dehydrogenase
2. Acquired Tolerance
Develops after repeated drug exposure. Has three subtypes:
A. Pharmacokinetic (Dispositional) Tolerance
- Reduced drug concentration at the site of action due to altered drug metabolism or distribution
- Most common mechanism: Enzyme induction of CYP450 system → accelerated hepatic metabolism → lower plasma drug levels for the same dose
- Examples:
- Barbiturates induce their own metabolism (autoinduction) and that of other drugs
- Rifampicin induces CYP enzymes → reduced levels of co-administered drugs
- Carbamazepine undergoes autoinduction - its own t½ shortens after starting therapy
- Ethanol (chronic use) induces CYP2E1
B. Pharmacodynamic (Cellular) Tolerance
- Drug concentration remains the same but the cellular/receptor response is diminished
- Most important and clinically significant mechanism
- Mechanisms include:
| Mechanism | Description | Example |
|---|---|---|
| Receptor downregulation | Prolonged agonist exposure reduces receptor density (number) | β₂-agonists (salbutamol) → reduced bronchodilation |
| Receptor desensitization | Receptor uncoupled from effector (G protein) without loss in number | Opioid receptors uncoupled from Gi proteins |
| Receptor internalization | Agonist-bound receptors internalized (endocytosed) and either recycled or degraded in lysosomes | Opioid receptor trafficking via β-arrestin/clathrin pathway |
| Increased NMDA receptor sensitivity | Counterregulatory upregulation of excitatory receptors | Opioid tolerance - NMDA contributes to reduced analgesia |
| Physiological adaptation | Compensatory changes in downstream signaling; counter-regulation by opposing systems | Opioids → reflex sympathetic activation; nitrates → reflex renin-angiotensin activation |
C. Learned (Behavioral) Tolerance
- Compensatory behavioral adaptations mask the overt effects of the drug
- The organism learns to function despite drug intoxication
- Example: Experienced alcohol drinkers appear less intoxicated at the same blood alcohol level due to learned behavioral compensation
Tachyphylaxis
- Rapid tolerance developing within minutes to hours of just a few doses
- Due to rapid receptor desensitization or depletion of mediator stores
- Examples:
- Ephedrine - acts by releasing noradrenaline from nerve terminals; stores become depleted rapidly
- Amphetamine - similar mechanism
- LSD - rapid tachyphylaxis within days; animals do not self-administer repeatedly
- Nitrates (short-acting) if used continuously
Cross-Tolerance
- Tolerance to one drug confers tolerance to pharmacologically related drugs acting at the same receptor
- Example: Morphine-tolerant patients show reduced response to all opioids (codeine, fentanyl, oxycodone)
- Important: Cross-tolerance between opioids is incomplete - different drugs may have different degrees of tolerance at the same receptor. This is exploited clinically in opioid rotation (switching to a different opioid at >50% below the calculated equianalgesic dose to restore analgesia)
Differential Tolerance
Not all effects of a drug develop tolerance at the same rate:
| Opioid Effect | Tolerance Development |
|---|---|
| Analgesia | Relatively faster |
| Sedation | Relatively faster |
| Nausea/vomiting | Relatively faster |
| Respiratory depression | Slower |
| Constipation | Slowest (often persists) |
| Miosis | Slowest (often persists) |
Clinical implication: An opioid-tolerant patient who requires higher doses for pain relief may still have significant respiratory depression risk - the analgesic effect and respiratory depressant effect do not develop tolerance equally.
Tolerance vs Dependence vs Addiction
These are distinct concepts that are frequently confused:
| Term | Definition |
|---|---|
| Tolerance | Rightward shift of dose-response curve; need more drug for same effect |
| Physical dependence | Physiological state of adaptation; withdrawal syndrome emerges on abrupt cessation or antagonist administration |
| Psychological dependence | Craving and compulsive drug-seeking behavior despite harm |
| Addiction | Chronic, treatable medical disease involving complex interactions of brain circuits, genetics, environment, and life experiences; compulsive use despite harmful consequences |
"Tolerance is not synonymous with dependence. The development of tolerance or physical dependence in no way implies that the patient is addicted to an opioid." - Miller's Anesthesia
Opioid Withdrawal (Physical Dependence)
On abrupt cessation of opioids, withdrawal is characterized by:
- Increased sympathetic activity: hypertension, tachycardia, diaphoresis
- GI effects: abdominal cramping, diarrhoea, nausea
- CNS: anxiety, restlessness, insomnia, yawning
- Musculoskeletal: myalgia, piloerection ("goose flesh" - hence "cold turkey")
Clinical Examples of Drug Tolerance
| Drug | Type of Tolerance | Mechanism | Clinical Impact |
|---|---|---|---|
| Opioids (morphine) | Pharmacodynamic | Receptor desensitization, downregulation, uncoupling from G proteins | Increasing dose needed for analgesia |
| Nitrates (GTN) | Pharmacodynamic | Tolerance develops within 24h of continuous use; involves free radical generation, reduced thiol groups, neurohormonal activation | Nitrate-free interval (8-12h/day) required |
| β₂-agonists (salbutamol) | Pharmacodynamic | β-arrestin-mediated receptor downregulation | Reduced bronchodilation with overuse |
| Benzodiazepines | Pharmacodynamic | GABA-A receptor uncoupling | Reduced anxiolytic/hypnotic effect |
| Barbiturates | Pharmacokinetic | CYP enzyme autoinduction | Shorter duration of effect |
| Ephedrine | Tachyphylaxis | Depletion of noradrenaline stores | Rapid loss of pressor effect with repeat doses |
| Alcohol | Pharmacokinetic + Learned | CYP2E1 induction + behavioral adaptation | Higher blood alcohol needed for same effect |
Mechanism of Nitrate Tolerance (Special Note)
Organic nitrates (GTN, isosorbide dinitrate) develop tolerance rapidly during continuous use:
- Mechanisms include: sulphydryl group depletion (needed for bioactivation), increased reactive oxygen species generation, reflex neurohormonal activation (renin-angiotensin, sympathetic), and plasma volume expansion
- Prevention: A nitrate-free interval of 8-12 hours/day (typically overnight for angina patches)
Reversal of Tolerance
- Rest (drug-free period): Receptor numbers recover; tolerance reverses
- Opioid rotation: Use incomplete cross-tolerance to restore analgesic sensitivity
- NMDA antagonists (e.g., ketamine): Can partially prevent or reverse opioid tolerance
- Buprenorphine: As a partial opioid agonist, used in managing opioid dependence precisely because it limits tolerance development
Sources: Katzung's Basic and Clinical Pharmacology, 16th Ed; Barash, Cullen & Stoelting's Clinical Anesthesia, 9th Ed; Miller's Anesthesia, 10th Ed
This is a shared conversation. Sign in to Orris to start your own chat.