Parkinson's disease in detail
Reading File
Finding Sources
Searching PubMed
"Parkinson disease"[MeSH Terms] AND management
Reading File
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Reading File
Loading Image
I now have comprehensive content from Harrison's and Katzung covering pathology, genetics, clinical features, and treatment. I have enough to write a thorough, well-organized response. Let me compile the full detailed answer.
Parkinson's Disease - A Comprehensive Overview
1. Definition and Epidemiology
Parkinson's disease (PD) is a progressive neurodegenerative disorder first described by James Parkinson in 1817 in his "Essay on the Shaking Palsy" - based on observations of only a handful of patients, some of whom he merely observed walking on the street.
- Estimated ~10.8 million people affected worldwide; expected to double within 20 years due to population aging
- Mean age of onset: ~60 years; cases can occur in the 20s, especially with pathogenic gene mutations
- Lifetime risk: ~3% in men, ~2% in women
- Frequency increases sharply with age
(Harrison's Principles of Internal Medicine 22E, p. 3537)
2. Pathology
The hallmark pathological features of PD are:
A. Substantia Nigra Depigmentation
Loss of the darkly pigmented dopaminergic neurons of the substantia nigra pars compacta (SNc) is the defining macroscopic change in PD. This is visible to the naked eye as pallor of the SNc.
Normal vs. PD brain - SNc depigmentation (Fig. 446-1A):
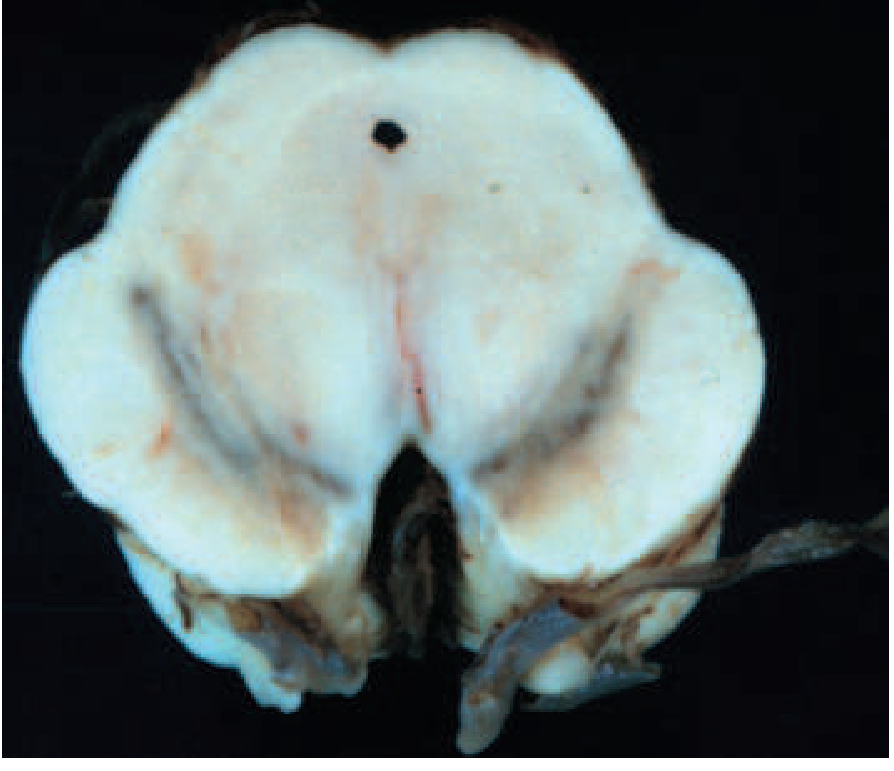
Left = normal SNc (dark pigmented). Right = PD brain (pale, depigmented SNc). - Harrison's, Fig. 446-1A
B. Lewy Bodies
Intraneuronal proteinaceous inclusions called Lewy bodies are the microscopic hallmark. They are round, eosinophilic inclusions with a dense core and pale halo, staining positive for α-synuclein and ubiquitin.
Histology - Lewy bodies (arrows) within melanized dopamine neurons in PD:
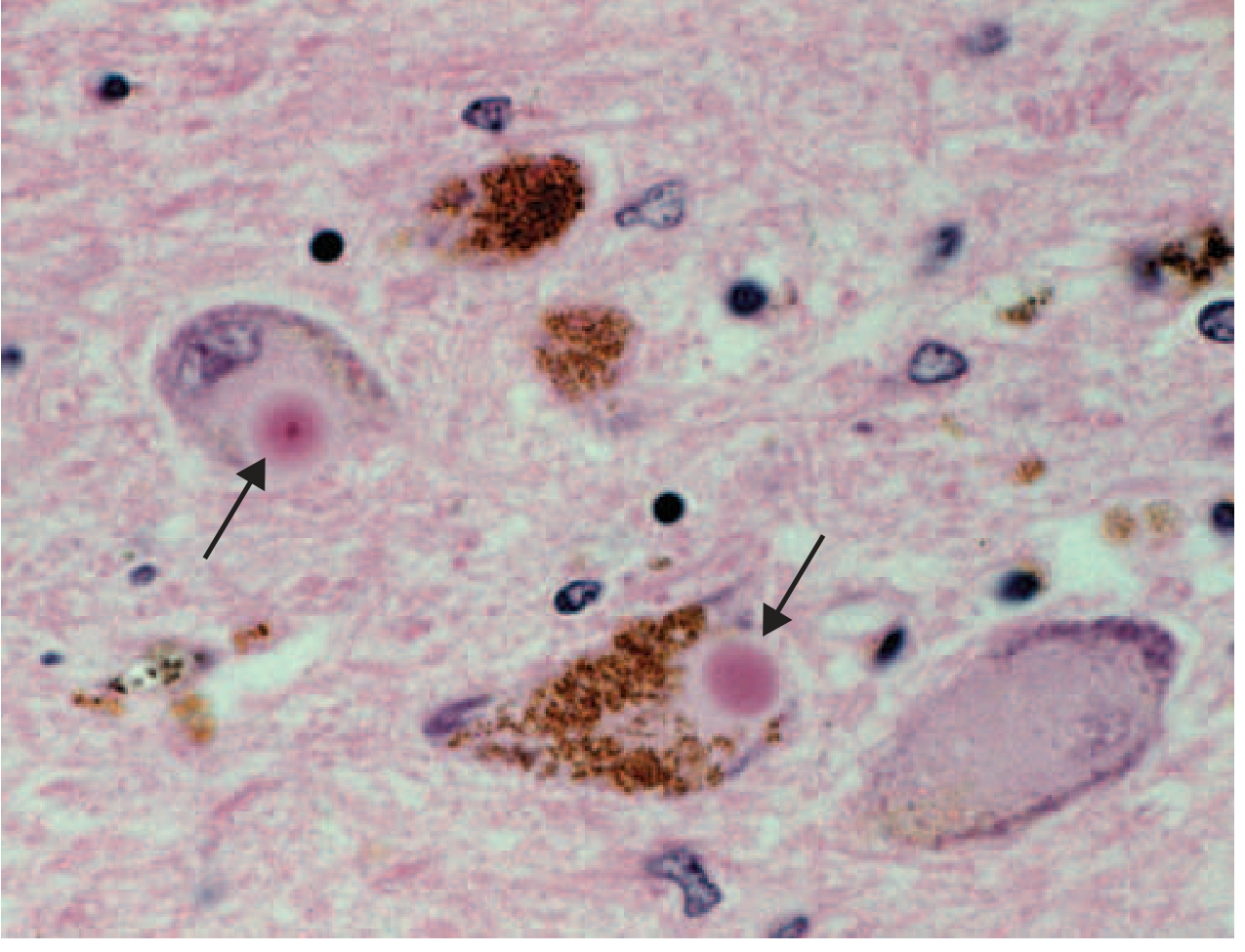
The pink circular inclusions (arrows) are Lewy bodies within brown-pigmented (melanized) nigral neurons. - Harrison's, Fig. 446-1C
C. Braak Staging
Pathology begins outside the SNc and spreads in a predictable pattern:
- Stage 1-2: Olfactory nucleus, dorsal motor nucleus of vagus, medulla/lower brainstem
- Stage 3-4: SNc affected (motor symptoms emerge), midbrain, mesocortex, thalamus
- Stage 5-6: Neocortex, widespread cortical involvement
This explains why anosmia, constipation, and REM sleep behavior disorder often precede motor symptoms by years.
(Katzung's Basic and Clinical Pharmacology 16e, p. 773; Harrison's, p. 3538)
D. Broader Neuronal Degeneration
PD is not purely a dopamine disorder. Lewy pathology also affects:
- Cholinergic neurons of the nucleus basalis of Meynert (NBM)
- Norepinephrine neurons of the locus coeruleus (LC)
- Serotonin neurons in raphe nuclei
- Olfactory system, cerebral hemispheres, spinal cord, peripheral autonomic nervous system
This widespread "nondopaminergic" degeneration accounts for the nonmotor features of PD. (Harrison's, p. 3538)
3. Pathogenesis
The pathogenesis involves a convergence of multiple mechanisms:
- Impaired protein degradation - ubiquitin-proteasome system dysfunction
- α-Synuclein misfolding and aggregation - the protein misfolds into toxic oligomers and protofibrils, seeding further aggregation (a "prion-like" mechanism; healthy fetal dopamine neurons transplanted into PD brains have been found to later develop Lewy pathology)
- Oxidative stress - dopamine metabolism generates reactive oxygen species; SNc neurons are particularly vulnerable
- Mitochondrial dysfunction - complex I inhibition (MPTP model)
- Neuroinflammation - microglial activation and inflammatory cascades
- Apoptosis of nigral neurons
(Katzung 16e, p. 773)
Environmental factors: Manganese dusts, carbon disulfide, carbon monoxide, lead exposure, farming, and vitamin D deficiency increase risk. Protective factors include cigarette smoking (paradoxically), coffee intake, NSAIDs, and high uric acid levels.
4. Genetics
Approximately 10-15% of cases have a recognized genetic cause. Key genes:
| Gene | Inheritance | Notes |
|---|---|---|
| SNCA (α-synuclein) | Autosomal dominant | Missense mutations + duplications/triplications; earlier onset, faster progression, prominent cognitive impairment. Triplication > duplication in severity |
| LRRK2 (leucine-rich repeat kinase 2, Chr 12) | Autosomal dominant | Most common known genetic cause of PD; clinically typical PD, slower progression; median age of onset 56 years |
| VPS35 | Autosomal dominant | Rare, clinically typical |
| GBA1 (glucocerebrosidase) | Risk factor | Faster progression, greater risk of cognitive impairment; heterozygous variants increase risk ~5-fold |
| Parkin (PARK2, Chr 6) | Autosomal recessive | Early-onset PD (juvenile/young adult); slower progression; Lewy bodies often absent |
| PINK1 | Autosomal recessive | Early-onset, slow progression |
| DJ-1 (PARK7) | Autosomal recessive | Rare, early onset |
| UCHL1 | Autosomal dominant | Rare |
The finding that SNCA duplication or triplication alone causes PD demonstrates that overproduction of normal α-synuclein protein is sufficient to cause disease.
(Harrison's 22e, p. 3538-3540; Katzung 16e, p. 773)
5. Clinical Features
Cardinal Motor Features (TRAP)
- Tremor - rest tremor, typically "pill-rolling" (4-6 Hz), diminishes with action, worsens at rest and with emotional stress
- Rigidity - "lead pipe" or "cogwheel" (if tremor superimposed), resistance throughout passive range of motion
- Akinesia/Bradykinesia - slowness of movement initiation and execution; the sine qua non required for diagnosis
- Postural instability - impaired righting reflexes; occurs later in disease
Other Motor Features
| Feature | Description |
|---|---|
| Micrographia | Progressively smaller handwriting |
| Masked facies (hypomimia) | Reduced facial expression, infrequent blinking |
| Hypophonia | Soft, monotone voice |
| Dysphagia | Present in 35% subjectively, up to 82% objectively |
| Freezing of gait | Sudden inability to initiate or continue steps |
| Festinating gait | Short shuffling steps, difficulty stopping |
| Stooped posture | Flexed trunk and limbs |
Nonmotor Features
These are often the most disabling in later disease and include:
Autonomic:
- Orthostatic hypotension
- Constipation (often a premotor symptom)
- Genitourinary dysfunction (urgency, incomplete emptying)
- Sexual dysfunction
- Seborrhea, sweating abnormalities
Neuropsychiatric:
- Depression (affects ~40%)
- Anxiety
- Apathy
- Psychosis (hallucinations - often visual - especially drug-related)
- Impulse control disorders (gambling, hypersexuality; linked to dopamine agonists)
- Cognitive impairment and dementia (Parkinson's disease dementia, PDD)
Sleep:
- REM sleep behavior disorder (RBD) - acting out dreams; can precede motor symptoms by years
- Excessive daytime sleepiness
- Insomnia
Sensory:
- Anosmia / hyposmia (often premotor)
- Pain, paresthesias
(Harrison's Table 446-1, p. 3537)
6. Diagnosis
PD remains a clinical diagnosis. The UK Parkinson's Disease Society Brain Bank Criteria are most widely used:
Step 1 - Diagnosis of Parkinsonism:
- Bradykinesia (required) PLUS at least one of: rest tremor, rigidity, postural instability
Step 2 - Exclusion criteria (features suggesting alternative diagnoses):
- History of repeated strokes, head injury, or encephalitis
- Oculomotor palsy (PSP), cerebellar signs (MSA), early severe autonomic failure
- Neuroleptic exposure at symptom onset
- Negative levodopa response
Step 3 - Supportive criteria (3 or more required for definite PD):
- Unilateral onset
- Rest tremor present
- Progressive disorder
- Persistent asymmetry
- Excellent levodopa response (70-100%)
- Levodopa-induced dyskinesia
-
10 years disease duration
Differential Diagnosis
| Category | Conditions |
|---|---|
| Atypical Parkinsonism | MSA-p, MSA-c, PSP (parkinsonian & Richardson's variants), CBS/CBD |
| Secondary Parkinsonism | Drug-induced (antipsychotics, metoclopramide), vascular, tumor, infection, NPH |
| Other Neurodegenerative | DLB (dementia with Lewy bodies), Wilson's disease, Huntington's, SCA3, NBIA |
Key "red flags" suggesting non-PD parkinsonism:
- Rapid progression
- Early falls (PSP)
- Early autonomic failure (MSA)
- Asymmetric cortical signs (CBS)
- Poor or no levodopa response
- Early dementia (DLB)
(Harrison's Table 446-2, p. 3538)
Investigations
- MRI brain: Mainly to exclude structural causes; usually normal in PD. MSA may show "hot cross bun" sign in pons
- DaTscan (SPECT with FP-CIT/ioflupane): Demonstrates reduced dopamine transporter in striatum; differentiates from essential tremor and drug-induced parkinsonism
- Genetic testing: Considered in early-onset (<50 years), positive family history
- Neuropsychological testing: For cognitive evaluation
- Autonomic testing: For orthostatic hypotension assessment
7. Treatment
PD is incurable and progressive, but pharmacologic treatment can relieve motor symptoms and improve quality of life for many years. Treatment is symptomatic; no agent has been proven neuroprotective in humans.
A. Levodopa (+ Carbidopa/Benserazide)
Levodopa remains the most effective drug for PD symptoms. It is converted to dopamine in the brain by DOPA decarboxylase.
- Combined with carbidopa (peripheral DOPA decarboxylase inhibitor) to prevent peripheral conversion, reducing dose requirements by ~75% and markedly reducing GI side effects
- Combination ratio: carbidopa/levodopa 10/100, 25/100, 25/250 (IR formulations); Rytary (extended-release capsules)
- Also available as Stalevo (carbidopa + levodopa + entacapone, a COMT inhibitor)
- Duodenal infusion (Duodopa/Lecigon) via PEG-J tube for advanced disease with motor fluctuations
Particularly effective for bradykinesia. About one-third of patients respond very well; ~one-third less well.
Adverse effects:
- GI: Nausea, vomiting (minimized by carbidopa co-administration; avoid phenothiazine antiemetics)
- Cardiovascular: Postural hypotension, cardiac arrhythmias
- CNS: Hallucinations, psychosis, confusion (especially in elderly)
- Motor complications (with long-term use):
- "Wearing off" (end-of-dose deterioration)
- "On-off" fluctuations
- Dyskinesias (involuntary movements at peak dose - choreiform)
- Diphasic dyskinesia
(Katzung 16e, p. 777-778; Harrison's 22e, p. 3544)
B. Dopamine Agonists
Act directly on dopamine receptors (primarily D2/D3). Longer half-lives than levodopa - reduce "wearing off" and motor fluctuations. Often used as initial monotherapy, especially in younger patients to delay levodopa and its motor complications.
| Drug | Class | Notes |
|---|---|---|
| Pramipexole (Mirapex) | Non-ergot | D2/D3 agonist; 0.125 mg TID start; renally excreted; extended-release available |
| Ropinirole (Requip) | Non-ergot | D2 agonist; 0.25 mg TID start; CYP1A2 metabolized; extended-release available |
| Rotigotine (Neupro) | Non-ergot | Transdermal patch; continuous delivery; 2 mg/24h start |
| Bromocriptine | Ergot | Older agent; now less used |
| Pergolide | Ergot | Associated with cardiac valvulopathy; largely withdrawn |
Adverse effects of all dopamine agonists:
- Nausea, vomiting (take with food)
- Orthostatic hypotension
- Peripheral edema
- Dyskinesias
- Hallucinations and psychosis (more than levodopa)
- Impulse control disorders (ICD) - pathological gambling, hypersexuality, compulsive eating, binge shopping - important to warn patients
- Excessive daytime sleepiness / sudden sleep attacks (driving warning)
(Katzung 16e, p. 780-782)
C. MAO-B Inhibitors
Inhibit monoamine oxidase type B, slowing dopamine breakdown in the brain.
| Drug | Dose | Notes |
|---|---|---|
| Selegiline (Deprenyl) | 5 mg BID | Also converted to amphetamine metabolites; possible mild neuroprotective effect debated |
| Rasagiline (Azilect) | 0.5-1 mg OD | No amphetamine metabolites; used as monotherapy or adjunct |
| Safinamide | 50-100 mg OD | Also has Na+ channel blocking and glutamate-modulating effects |
Mild symptomatic benefit; useful in early disease as monotherapy. Also used as adjunct to levodopa to reduce "off" time.
D. COMT Inhibitors
Catechol-O-methyltransferase inhibitors block peripheral and central degradation of levodopa/dopamine, prolonging levodopa's effect.
| Drug | Notes |
|---|---|
| Entacapone | Peripheral COMT inhibitor; given with each levodopa dose; available combined as Stalevo |
| Tolcapone | Both peripheral and central COMT inhibition; more potent; requires LFT monitoring (hepatotoxicity risk) |
| Opicapone | Once-daily peripheral COMT inhibitor; newer agent |
Used as adjuncts to reduce "off" time in fluctuating patients.
E. Amantadine
- NMDA receptor antagonist; mild dopaminergic and anticholinergic properties
- Main role: Only oral drug with evidence for reducing levodopa-induced dyskinesia without worsening parkinsonism
- Side effects: Livedo reticularis, leg edema, cognitive impairment, hallucinations
- Must be tapered on withdrawal (withdrawal syndrome)
- Extended-release formulation (GOCOVRI) available
(Harrison's 22e, p. 3546)
F. Anticholinergics
- Trihexyphenidyl, benztropine mesylate
- Historically first-line, now largely superseded by levodopa
- Main benefit: tremor
- Avoid in elderly - cognitive impairment, urinary retention, glaucoma, constipation
- Still useful in selected younger patients with severe, refractory tremor
G. Adenosine A2A Receptor Antagonist
- Istradefylline (Nourianz) - the only approved A2A antagonist
- Adjunct therapy for "off" episodes in adults on levodopa/carbidopa
- Mechanism: Blocks A2A receptors that co-localize with D2 receptors on indirect pathway neurons, restoring balance in basal ganglia-thalamocortical circuit without increasing levodopa dose
- Interestingly, caffeine is also an A2A antagonist (possibly explaining epidemiologic protection with coffee consumption)
(Harrison's 22e, p. 3546)
H. Drug Treatment Table Summary
| Drug Class | Agents | Main Use |
|---|---|---|
| Levodopa combinations | Carbidopa/levodopa (Sinemet, Rytary, Duodopa) | First-line for moderate-severe motor symptoms |
| Dopamine agonists | Pramipexole, ropinirole, rotigotine | Early-onset PD, adjunct in advanced PD |
| MAO-B inhibitors | Rasagiline, selegiline, safinamide | Early disease monotherapy, adjunct |
| COMT inhibitors | Entacapone, opicapone, tolcapone | Adjunct to reduce "off" time |
| NMDA antagonist | Amantadine | Anti-dyskinesia; early mild motor symptoms |
| Anticholinergics | Trihexyphenidyl, benztropine | Tremor (younger patients only) |
| A2A antagonists | Istradefylline | Adjunct for "off" time |
(Harrison's Table 446-5, p. 3547)
8. Surgical Treatment: Deep Brain Stimulation (DBS)
DBS is the main surgical treatment for advanced PD with motor complications not adequately controlled by medications. High-frequency electrical stimulation is delivered via implanted electrodes.
Targets:
- Subthalamic nucleus (STN) DBS - larger benefit in medication-off state; allows greater dopaminergic medication reduction; possibly slightly higher neuropsychiatric risk
- Globus pallidus internus (GPi) DBS - better dyskinesia suppression; better for "brittle" dyskinesia (dyskinesia at low doses); better long-term flexibility; relatively safer neuropsychiatric profile
Both STN and GPi DBS show similar overall motor outcomes. STN DBS allows greater medication reduction; GPi DBS may be preferred when dyskinesia management or neuropsychiatric stability is the priority.
Patient selection:
- Advanced PD with disabling motor fluctuations or dyskinesia
- Good levodopa responsiveness (predicts DBS response)
- Cognitively intact (relative contraindication if dementia)
- No significant psychiatric comorbidity
- Generally <75 years (relative)
(Bradley and Daroff's Neurology in Clinical Practice; Harrison's 22e, p. 3548)
9. Nonpharmacological Management
- Exercise - strong evidence for improving motor and nonmotor symptoms; a 2025 network meta-analysis (PMID 39880702) found optimized exercise type and dose significantly improves motor function
- Physiotherapy - gait training, balance, fall prevention
- Occupational therapy - adaptive equipment, ADL strategies
- Speech therapy - Lee Silverman Voice Treatment (LSVT) for hypophonia; swallowing evaluation and management
- Psychological support - CBT for depression and anxiety; a 2025 meta-analysis (PMID 40568748) addressed anxiety management specifically
- Virtual reality therapy - 2025 systematic review (PMID 40142335) showed VR + conventional therapy significantly improves balance compared to conventional therapy alone
- Dietary management - protein redistribution (levodopa absorption impaired by amino acids); adequate hydration and fiber for constipation
- Sleep hygiene - management of RBD (clonazepam, melatonin), sleepiness, insomnia
10. Complications and Disease Progression
| Stage | Features |
|---|---|
| Early | Unilateral tremor/rigidity/bradykinesia; good levodopa response; minimal disability |
| Moderate | Bilateral involvement; gait difficulty; some falls; ADL partially impaired |
| Advanced | Postural instability; frequent falls; motor fluctuations; dyskinesias; nonmotor burden |
| Late | Wheelchair-bound; dementia common; aspiration pneumonia risk; nursing home care |
Major causes of morbidity/mortality:
- Aspiration pneumonia - dysphagia present in up to 82% objectively; silent aspiration in 15-33%; ~10-fold increased respiratory infection risk in those with drooling and silent aspiration (Bradley and Daroff's Neurology)
- Falls and fractures
- Dementia (Parkinson's disease dementia / overlap with DLB)
- Psychosis (often drug-related)
- Autonomic failure (severe orthostatic hypotension, urinary dysfunction)
11. Key Distinctions: PD vs. Parkinsonism
| Feature | Idiopathic PD | Drug-induced | PSP | MSA |
|---|---|---|---|---|
| Onset | Insidious, unilateral | After neuroleptic exposure | Insidious, axial > limb | Insidious |
| Tremor | Classic rest tremor | Variable | Uncommon | Uncommon |
| Levodopa response | Excellent (70-100%) | Partial or none | Poor | Poor to moderate |
| Early falls | Late | N/A | Early (backwards) | Early |
| Eye movement | Normal early | Normal | Vertical gaze palsy | Variable |
| MRI | Normal | Normal | Midbrain "hummingbird" sign | Pons "hot cross bun" |
| Progression | Slow-moderate | Reverses on drug withdrawal | Rapid | Moderate-rapid |
Summary
Parkinson's disease is the second most common neurodegenerative disorder, characterized pathologically by dopaminergic neuronal loss in the SNc, reduced striatal dopamine, and Lewy body (α-synuclein) deposition. Its cardinal motor features - bradykinesia, rest tremor, rigidity, and postural instability - emerge at Braak stage 3, but extensive nonmotor and premotor features (anosmia, constipation, RBD, depression) reflect the wider spread of Lewy pathology. Treatment remains symptomatic: levodopa/carbidopa is the most effective agent, supplemented by dopamine agonists, MAO-B inhibitors, COMT inhibitors, amantadine, and DBS for advanced disease. No neuroprotective therapy is yet proven in humans.
Sources:
- Harrison's Principles of Internal Medicine 22E (2025), Ch. 446 "Parkinson's Disease and Related Disorders"
- Katzung's Basic and Clinical Pharmacology, 16th Ed., Ch. 28 "Pharmacological Management of Parkinsonism & Other Movement Disorders"
- Bradley and Daroff's Neurology in Clinical Practice, Ch. 38 (Deep Brain Stimulation in PD)
- Campbell-Walsh Urology; Sleisenger and Fordtran's GI and Liver Disease (autonomic/GI aspects)
This is a shared conversation. Sign in to Orris to start your own chat.