Explain about cardiacmyopathy from basics
Reading File
Finding Sources
Searching the Web
cardiomyopathy types classification pathophysiology overview 2024
Reading File
Reading File
Reading File
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Finding Sources
Loading Image
Finding Sources
Loading Image
Reading File
Reading File
I now have enough comprehensive content to write a thorough response. Let me compile everything:
Cardiomyopathy: From Basics to Clinical Management
What Is Cardiomyopathy?
Cardiomyopathy refers to diseases of the heart muscle (myocardium) that are intrinsic to the cardiac muscle itself - not caused by coronary artery disease, valvular disease, hypertension, or congenital defects. The word breaks down simply: cardio (heart) + myo (muscle) + pathy (disease).
The WHO/ESC definition has evolved over decades. The current framework classifies them as:
- Primary cardiomyopathies - predominantly involve the heart muscle (genetic, mixed, or acquired)
- Secondary cardiomyopathies - heart muscle involvement as part of a systemic disease
Source: Goldman-Cecil Medicine, Fuster and Hurst's The Heart 15e, Harrison's Principles 22e
Classification by Pathophysiology
There are four major types, classified by ventricular morphology and function:
| Type | Ejection Fraction | Core Dysfunction | Key Mechanism |
|---|---|---|---|
| Dilated (DCM) | <40% | Systolic (contractile) | Ventricular dilation, weak pump |
| Hypertrophic (HCM) | 50-80% | Diastolic (relaxation) | Abnormal thickening, stiff ventricle |
| Restrictive (RCM) | Normal/near-normal | Diastolic (filling) | Stiff, non-compliant myocardium |
| Arrhythmogenic (ARVC) | Variable (RV affected) | Arrhythmia + structural | Fibro-fatty replacement of RV |
DCM accounts for approximately 90% of all cardiomyopathy cases. - Robbins, Cotran & Kumar Pathologic Basis of Disease
1. Dilated Cardiomyopathy (DCM)
Definition
DCM is defined by dilation and impaired systolic function of the left ventricle (or both ventricles), in the absence of coronary artery disease, valvular abnormalities, or pericardial disease. Prevalence in adults is approximately 1 in 250.
Causes
Genetic (20-50% of cases):
- Autosomal dominant inheritance is the predominant pattern
-
50 genes implicated; most common are mutations in titin (TTN) - accounting for ~25% of familial cases
- Also: beta-myosin heavy chain (MYH7), cardiac troponin T, alpha-myosin heavy chain
- The mutations primarily cause loss of function in cytoskeletal or sarcomere proteins (contrast with HCM which is gain-of-function)
- X-linked: mutations in dystrophin (as in Duchenne/Becker muscular dystrophy)
Acquired causes:
- Infectious myocarditis (viral - coxsackievirus B, parvovirus B19, adenovirus)
- Alcohol - toxic effect of alcohol and acetaldehyde on myocardium
- Chemotherapy (e.g., doxorubicin/anthracyclines)
- Peripartum/pregnancy
- Radiation therapy
- Nutritional deficiencies (thiamine, selenium)
- Iron overload (hemochromatosis)
- Cocaine, tachycardia-mediated, sarcoidosis
Pathophysiology
The heart dilates to compensate for poor contractility - a form of eccentric remodeling. The result:
- Reduced stroke volume and cardiac output
- Elevated filling pressures → pulmonary congestion
- Ventricular dilation promotes dispersion of depolarization/repolarization → risk of ventricular arrhythmias and sudden death
Clinical Features
- Progressive dyspnea (exertional, then at rest)
- Fatigue, reduced exercise tolerance
- Signs of heart failure: S3 gallop, displaced apex beat, peripheral edema, elevated JVP
- Mitral/tricuspid regurgitation due to annular dilation
- Embolic events (mural thrombus in dilated ventricle)
Investigations
- Echo: Dilated LV with globally reduced EF, wall motion abnormality
- ECG: Left bundle branch block, non-specific ST changes, arrhythmias
- BNP/NT-proBNP: Elevated, reflects wall stress
- Genetics: TTN mutation screening in familial cases
2. Hypertrophic Cardiomyopathy (HCM)
Definition
HCM is unexplained left ventricular hypertrophy (LV wall thickness ≥15 mm) in the absence of a causative hemodynamic factor (hypertension, aortic stenosis). Prevalence ~1:500. It is the most common cardiovascular cause of sudden death in young athletes, accounting for one-third of such events.
Echocardiogram of HCM
Below is a classic echocardiographic appearance of HCM - note the thick interventricular septum and anterior motion of the mitral valve (MV) toward it:
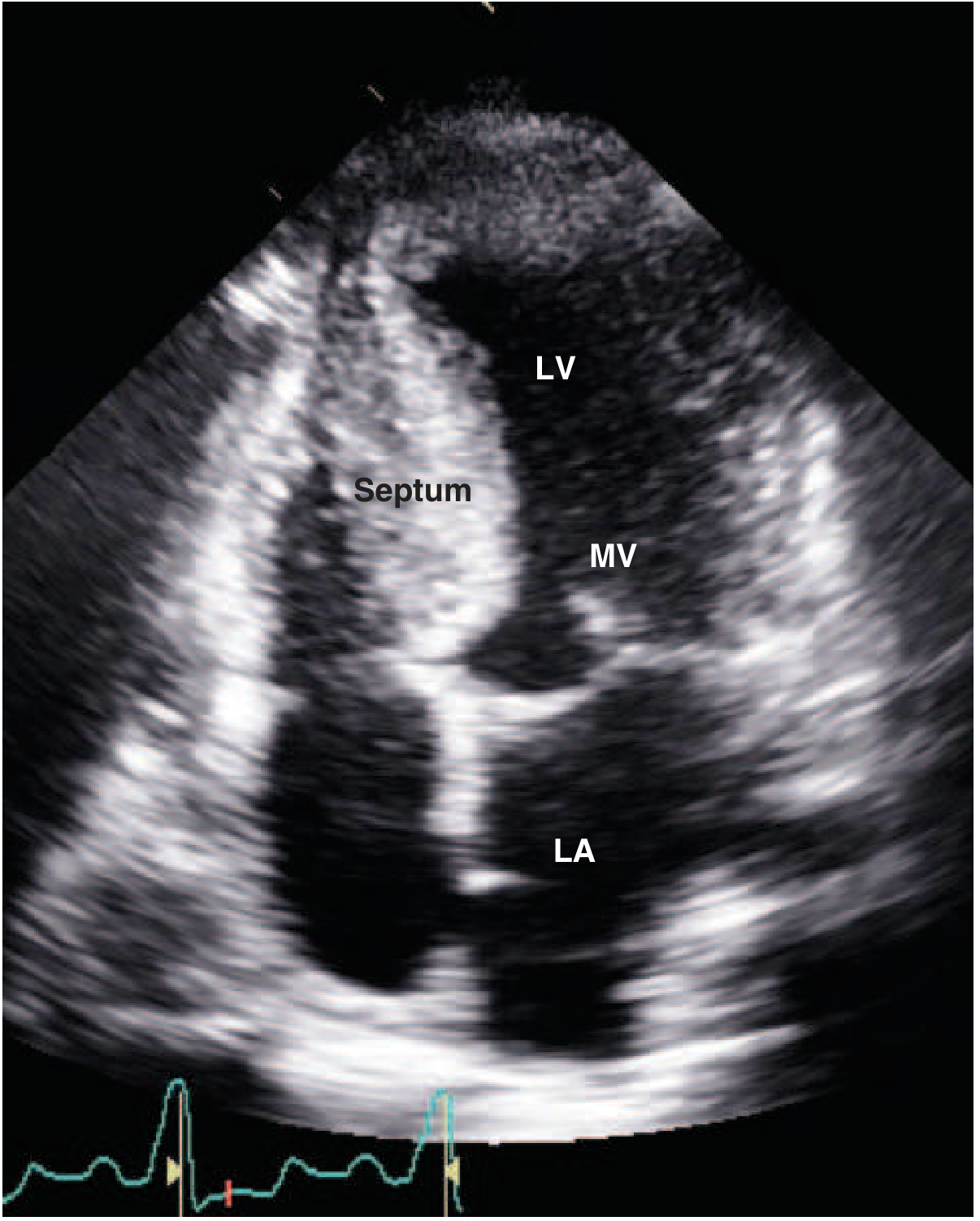
Genetics
- ~40-50% have a sarcomere gene variant; ~80% of those involve MYH7 (beta-myosin heavy chain) or MYBPC3 (myosin-binding protein C)
-
1500 genetic variants identified across >9 genes
- Autosomal dominant, but with age-dependent and incomplete penetrance
- The hypertrophic phenotype is rarely present at birth; usually develops in adolescence/young adulthood
- The mutations cause a gain of function at the sarcomere level - enhanced calcium sensitivity, maximal force generation, and ATPase activity - leading to abnormal energetics and impaired relaxation
Histology of HCM
The hallmark microscopic finding is myocyte disarray - myofibrils arranged in a chaotic, swirling pattern rather than the normal parallel arrangement:
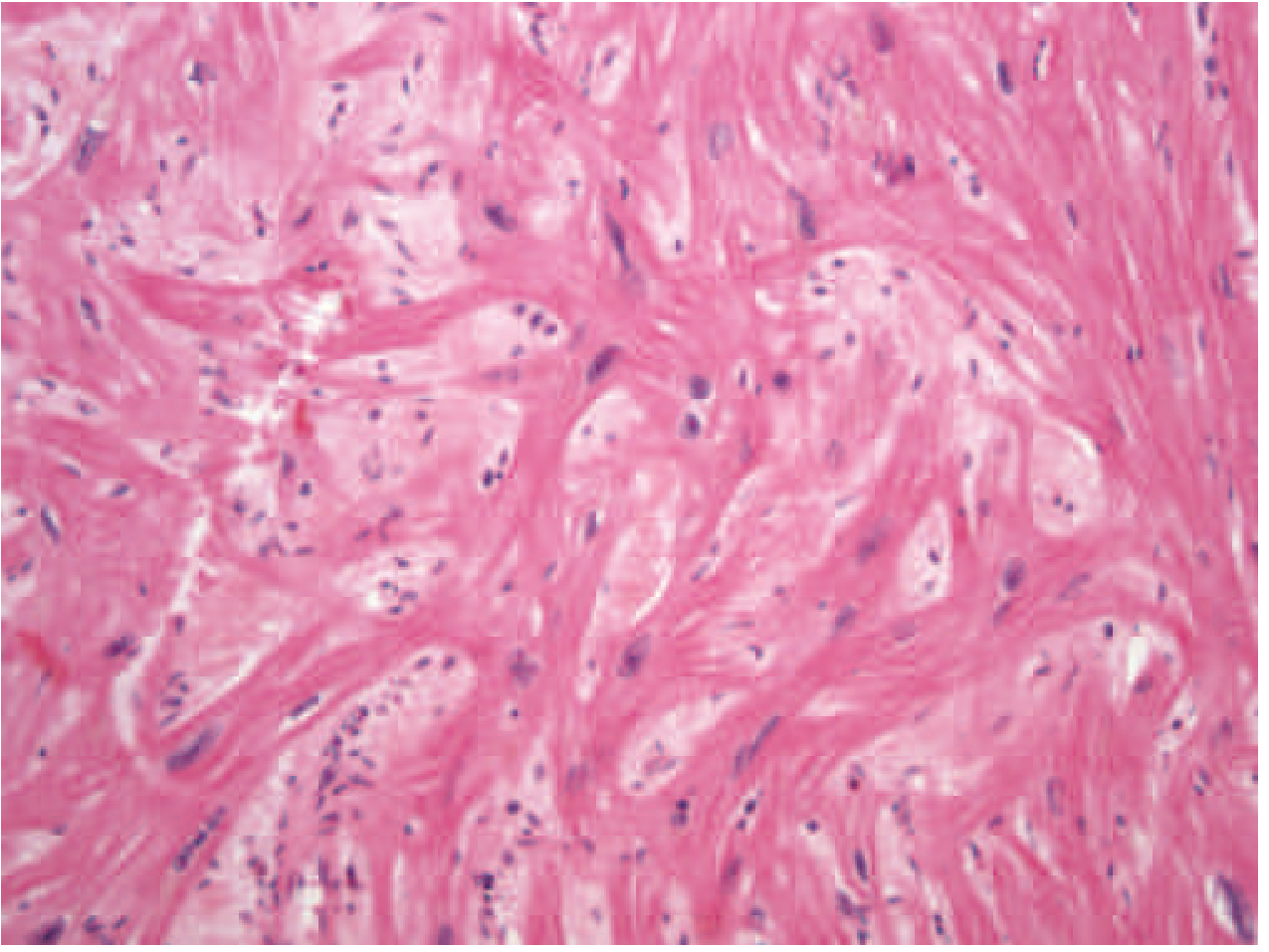
Pathophysiology
- Diastolic dysfunction is the primary problem - the stiff, hypertrophied LV cannot relax properly, impairing filling
- LVOT obstruction is present in ~30% at rest and can be provoked by exercise in another ~30%
- Obstruction mechanism: drag forces push the anteriorly displaced anterior mitral leaflet into the hypertrophied septum during systole (SAM - systolic anterior motion) → outflow tract narrowing + posteriorly directed mitral regurgitation
- Conditions that reduce preload (dehydration) or afterload (vasodilators) worsen obstruction
- The classic murmur: harsh, late-peaking systolic ejection murmur at the LLSB, increased by Valsalva and standing (decreased ventricular volume worsens obstruction), decreased by squatting
Clinical Features
- Exertional dyspnea, angina, syncope (especially with exertion)
- Sudden cardiac death (most feared, especially in young athletes)
- Palpitations (AF, VT)
- Family history in ~50%
- Risk of sudden death increases ~1% per year
HCM vs. Athlete's Heart
| Feature | HCM | Athlete's Heart |
|---|---|---|
| LV cavity size | Normal/small | Mildly dilated |
| Wall thickness regression on detraining | No | Yes |
| Diastolic function | Abnormal | Normal |
| VO2max | Normal/reduced | >50 mL/kg/min |
| Family history | Often positive | Negative |
3. Restrictive Cardiomyopathy (RCM)
Definition
RCM is characterized by a stiff, non-compliant myocardium that restricts ventricular filling (diastolic dysfunction), while systolic function is often preserved initially. It is the least common of the three classic types.
Causes
- Infiltrative: Amyloidosis (AL or ATTR type - most important), sarcoidosis
- Storage diseases: Hemochromatosis (iron overload), Fabry disease, glycogen storage diseases
- Fibrotic: Post-radiation fibrosis, post-cardiac surgery, hypereosinophilic syndrome
- Endomyocardial fibrosis (EMF): A tropical disease causing obliteration of ventricular apices
- Löeffler endocarditis: Eosinophilic infiltration
Key: Cardiac Amyloidosis
Amyloid deposition in the myocardium is described as a "toxic-infiltrative cardiomyopathy" - two mechanisms operate: (1) interstitial/perivascular amyloid fibril deposition disrupting tissue architecture and causing microvascular dysfunction, and (2) direct cardiotoxic effects. The ECG classically shows low voltage with a pseudo-infarct pattern (Q waves) - a paradox in a thick heart.
Clinical Features
- Dyspnea, fatigue, peripheral edema
- Elevated JVP with Kussmaul's sign (JVP rises on inspiration, opposite to normal)
- S3 and S4 gallop
- Right heart failure may be prominent
- Must be distinguished from constrictive pericarditis (both cause diastolic restriction)
4. Arrhythmogenic (Right Ventricular) Cardiomyopathy (ARVC)
ARVC is a hereditary disease characterized by fibro-fatty replacement of right ventricular myocardium. Key features:
- Right-sided heart failure
- Ventricular arrhythmias with LBBB morphology (originating from RV)
- Syncope and sudden cardiac death
- ECG: epsilon wave (a small notch after the QRS in V1-V3), T-wave inversions in right precordial leads
- Caused by mutations in desmosomal proteins (plakophilin-2 most common, desmoplakin, desmoglein-2)
- ICD implantation recommended for high-risk patients
Comparing All Four Types: Summary Table
| Feature | DCM | HCM | RCM | ARVC |
|---|---|---|---|---|
| Ventricular size | Dilated (bilateral) | Normal/small LV | Normal/mildly enlarged | RV enlarged |
| Wall thickness | Normal/thin | Increased | Normal | Normal/thin |
| Systolic function | Reduced (EF <40%) | Hyperdynamic | Normal/near-normal | RV reduced |
| Diastolic function | Normal/reduced | Impaired (primary) | Severely impaired | Variable |
| Key mutation | Titin (TTN), dystrophin | MYH7, MYBPC3 | Desmosomal, TTR (amyloid) | DSP, PKP2 |
| Inheritance | AD, X-linked | AD | Variable | AD |
| Most feared complication | Heart failure, SCD | SCD in young athletes | Progression to HF | SCD |
Approach to Diagnosis
- History: Family history of SCD, heart failure, symptoms (dyspnea, syncope, palpitations)
- Physical exam: Murmurs, S3/S4, signs of heart failure
- ECG: LBBB in DCM; LVH + deep T inversions in HCM; epsilon wave in ARVC; low voltage + pseudo-infarct in amyloid
- Echocardiography: First-line imaging - defines anatomy, EF, wall motion, LVOT gradient
- Cardiac MRI: Gold standard for tissue characterization (fibrosis by late gadolinium enhancement, fatty infiltration in ARVC)
- Genetic testing: Recommended in familial disease; guides family screening
- Endomyocardial biopsy: For suspected infiltrative disease (amyloid, sarcoid, myocarditis)
- Lab: BNP, troponin, iron studies, TTR gene sequencing
General Treatment Principles
DCM / Heart Failure with Reduced EF
The "Fantastic Four" guideline-directed medical therapy (GDMT):
- ACE inhibitors / ARBs / ARNi (sacubitril-valsartan) - reduce afterload and remodeling
- Beta-blockers (carvedilol, metoprolol succinate, bisoprolol) - reduce heart rate, improve remodeling
- Mineralocorticoid receptor antagonists (spironolactone, eplerenone) - reduce fibrosis
- SGLT2 inhibitors (dapagliflozin, empagliflozin) - reduce hospitalizations and mortality
Device therapy: ICD for EF <35% (SCD prevention); CRT for EF <35% + LBBB (resynchronization).
HCM
- Avoid vasodilators, diuretics (worsen obstruction)
- Beta-blockers or calcium channel blockers (verapamil) for symptoms
- Disopyramide for refractory LVOT obstruction
- Mavacamten (cardiac myosin inhibitor) - new targeted therapy, reduces LVOT gradient
- Septal reduction therapy (surgical myectomy or alcohol septal ablation) for severely symptomatic obstruction
- ICD implantation for high-risk patients (SCD risk factors: wall thickness ≥30 mm, VT, family history of SCD, syncope)
RCM
- Treat the underlying cause (chelation for hemochromatosis, steroids for sarcoid)
- Diuretics cautiously (preload-dependent filling)
- Heart transplantation for end-stage disease
ARVC
- ICD (primary prevention in high-risk)
- Beta-blockers, sotalol, amiodarone for arrhythmia suppression
- Avoid intense competitive sports
- Catheter ablation for refractory VT
Myocarditis and Its Link to DCM
Myocarditis is myocardial damage caused by inflammatory infiltrates secondary to viral infections (most common in the US: coxsackievirus B, adenovirus, parvovirus B19) or immune reactions. Clinically it can be asymptomatic, cause acute heart failure, or evolve into DCM - this is one of the most important links, as the viral trigger may "burn out" leaving behind a dilated, poorly functioning heart. - Robbins, Cotran & Kumar Pathologic Basis of Disease
Key Points to Remember
- Cardiomyopathy = intrinsic heart muscle disease - not from ischemia, valve disease, or hypertension
- DCM: dilated + weak pump (systolic failure); most common type
- HCM: thick + stiff (diastolic failure, outflow obstruction); #1 cause of sudden death in young athletes
- RCM: stiff + non-compliant (filling problem); least common; think amyloid
- ARVC: fibro-fatty RV replacement; arrhythmia and SCD in young adults
- Genetics plays a central role in all four types - family screening is mandatory
- Echocardiogram is first-line; cardiac MRI for tissue characterization
For a great visual overview, this educational video by Armando Hasudungan covers the key concepts well:
Sources: Robbins, Cotran & Kumar Pathologic Basis of Disease (10e); Harrison's Principles of Internal Medicine (22e, 2025); Goldman-Cecil Medicine International Edition; Fuster and Hurst's The Heart (15e)
This is a shared conversation. Sign in to Orris to start your own chat.