Amylodosis pathology University answer pattern
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Finding Sources
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
I now have comprehensive, exam-quality content from Robbins. Let me compose the full university-pattern answer:
AMYLOIDOSIS - University Examination Answer
(Robbins, Cotran & Kumar Pathologic Basis of Disease | Robbins & Kumar Basic Pathology)
DEFINITION
Amyloidosis is a generic term for a group of disorders in which normally soluble proteins misfold and aggregate into insoluble fibrils that deposit in extracellular tissues, causing progressive organ damage and functional compromise.
The name "amyloid" derives from the mistaken early belief that deposits resembled starch (amylose), based on staining reactions with iodine. The deposits are unrelated to starch.
PROPERTIES OF AMYLOID
Physical Nature
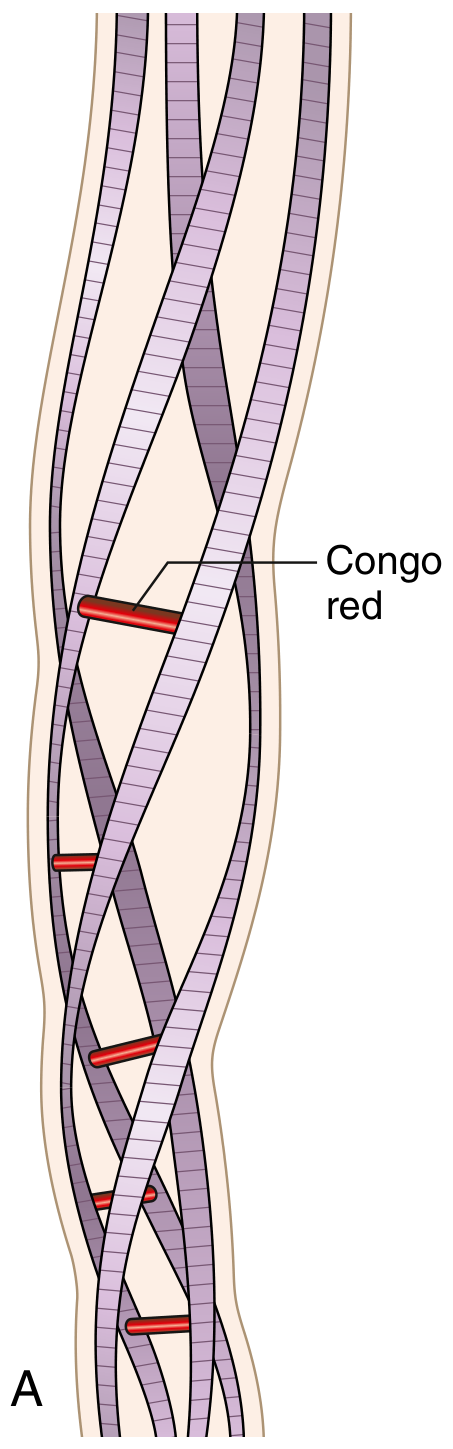
- By electron microscopy: continuous, non-branching fibrils, diameter 8-10 nm
- Each fibril consists of stacked protofilaments in a beta (β)-pleated sheet conformation
- This β-pleated sheet conformation is responsible for all characteristic staining properties
Chemical Composition
- ~95% fibril proteins (amyloidogenic proteins)
- ~5% serum amyloid P (SAP) component + glycoproteins (heparan sulfate, dermatan sulfate proteoglycans)
Staining Characteristics (HIGH YIELD)
| Stain | Appearance |
|---|---|
| H&E | Amorphous, eosinophilic, hyaline, extracellular substance |
| Congo red (routine light) | Pink/salmon-red color |
| Congo red (polarized light) | Apple-green birefringence - PATHOGNOMONIC |
| Crystal violet | Metachromasia (purple) |
| Thioflavin T (fluorescence) | Yellow-green fluorescence |
| PAS | Weakly positive, diastase-resistant |
The apple-green birefringence under polarized light with Congo red is the GOLD STANDARD for diagnosis.
PATHOGENESIS
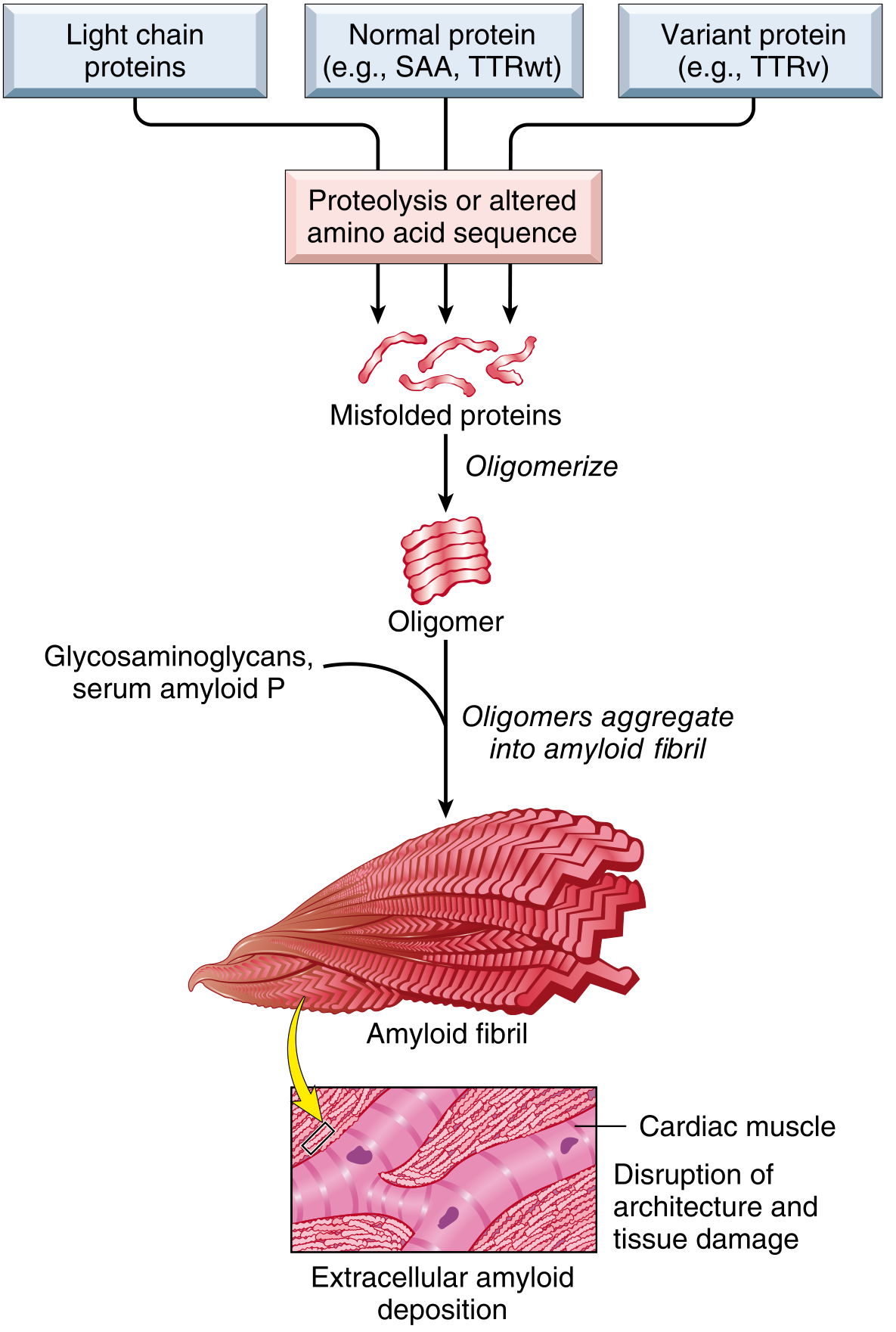
Amyloidosis results from failure of protein quality control mechanisms:
- Normally, intracellular misfolded proteins are degraded in proteasomes
- Extracellular protein aggregates are cleared by macrophages
- In amyloidosis, both mechanisms fail → fibrillar protein accumulation outside cells
Two categories of amyloidogenic proteins:
- Normal proteins with inherent tendency to self-associate, particularly when overproduced (e.g., SAA in chronic inflammation, transthyretin wild-type)
- Mutant/variant proteins structurally prone to misfolding (e.g., mutant transthyretin in familial amyloidosis)
Organ dysfunction is mainly caused by:
- Abnormal tissue architecture from extracellular deposits
- Activation of ROS-producing signaling pathways
- Defective calcium homeostasis
CLASSIFICATION (Table - University Essential)
| Type | Fibril Protein | Precursor | Associated Condition |
|---|---|---|---|
| AL (Primary) | Immunoglobulin light chain fragments (mainly λ) | Plasma cell light chains | Multiple myeloma, monoclonal gammopathy |
| AA (Secondary/Reactive) | AA protein (8500-dalton) | Serum amyloid A (SAA) from liver | Chronic inflammation: RA, Crohn's, TB, osteomyelitis, heroin skin infections |
| ATTR (Transthyretin) | Transthyretin (TTR) | TTR protein | Wild-type (senile systemic); Mutant (familial neuropathy/cardiomyopathy) |
| Aβ2M (Dialysis-related) | β2-microglobulin | β2-microglobulin (filtered by kidney) | Long-term hemodialysis |
| Aβ (Alzheimer's) | Aβ peptide (4000 daltons) | Amyloid precursor protein (APP) | Alzheimer's disease, Down syndrome |
| Familial/Hereditary | Mutant TTR, fibrinogen, lysozyme | Various gene mutations | Familial Mediterranean fever (AA), FAP |
CLASSIFICATION BY DISTRIBUTION
Systemic (Generalized) Amyloidosis
- Involves multiple organs
- Subtypes: Primary (AL), Reactive/Secondary (AA), Heredofamilial (ATTR mutant), Dialysis-related (Aβ2M)
Localized Amyloidosis
- Deposits limited to single organ/tissue
- Examples: Alzheimer's (brain), medullary thyroid carcinoma (calcitonin-derived), senile cardiac, cutaneous, endocrine tumors
MAJOR CLINICAL FORMS (Detailed)
1. AL Amyloidosis (Primary)
- Most common form - ~2000-3000 new cases/year in the USA
- Clonal plasma cell proliferation secreting abnormal Ig light chains
- λ chains 6x more likely to deposit as amyloid than κ chains
- 5-15% of multiple myeloma patients develop systemic amyloidosis
- Most cases: no overt B-cell neoplasm, but monoclonal Ig or free light chains (Bence Jones protein) detectable in blood/urine
- Organs affected: kidney (most common - nephrotic syndrome), heart, liver, spleen, peripheral nerves, tongue (macroglossia), GI tract
2. AA Amyloidosis (Reactive/Secondary)
- Composed of AA protein derived by proteolysis of serum amyloid-associated (SAA) protein (synthesized by liver)
- SAA synthesis stimulated by IL-6 and IL-1 (acute-phase response cytokines) during chronic inflammation
- Associated conditions:
- Chronic infections: TB, osteomyelitis, bronchiectasis (now less common due to antibiotics)
- Rheumatoid arthritis (most common current association) - occurs in ~3% of RA patients
- Inflammatory bowel disease (Crohn's > UC)
- Ankylosing spondylitis
- Chronic IV/subcutaneous heroin use (skin infections)
- Cancers: renal cell carcinoma, Hodgkin lymphoma
- Organs affected: kidney (proteinuria/nephrotic syndrome), liver, spleen, adrenals
3. Heredofamilial / Familial Amyloidosis
- Familial Mediterranean Fever (FMF): autosomal recessive; Armenian, Sephardic Jewish, Arabic populations; amyloid is AA type; mutation in MEFV gene (encodes pyrin/marenostrin); presents with recurrent inflammatory attacks (peritonitis, pleuritis, arthritis) + amyloidosis
- Familial Amyloid Polyneuropathy (FAP): autosomal dominant; mutant TTR (V30M most common, occurs in 30-45%); presents with peripheral neuropathy + cardiomyopathy
- Senile Systemic Amyloidosis: wild-type (normal) TTR deposits in heart in elderly patients (>70 years) - slowly progressive restrictive cardiomyopathy
4. Dialysis-Associated Amyloidosis
- β2-microglobulin, normally filtered by the kidney, accumulates in patients on long-term hemodialysis
- Deposits in joints, synovium, tendons, carpal tunnel (carpal tunnel syndrome)
MORPHOLOGY (Gross and Microscopic)
Gross Appearance
- Affected organs are enlarged and firm ("lardaceous" = waxy, bacon-like appearance)
- Classic test: Lugol's iodine → mahogany-brown; then H2SO4 → blue-violet color ("amyloid reaction")
Microscopic Appearance
H&E: amorphous, homogeneous, glassy eosinophilic material in extracellular spaces
Kidney (most commonly affected in systemic amyloidosis):
- Deposits initially in mesangium, then capillary walls of glomeruli
- Later: tubular BM and vessels
- Results in nephrotic syndrome → chronic renal failure
Spleen - two patterns:
- Sago spleen: deposits in splenic follicles (white pulp) → small, tapioca-like granules on cut surface
- Lardaceous spleen: deposits in splenic sinuses (red pulp) → large, irregular, waxy areas
Liver:
- Deposits in space of Disse (between hepatocytes and sinusoidal endothelium)
- Progressive compression of hepatocytes → hepatomegaly (mild dysfunction; jaundice rare)
Heart:
- Deposits in interstitium between myocardial fibers → restrictive cardiomyopathy
- Subendocardial deposits → "senile cardiac amyloidosis" pattern
Tongue:
- Macroglossia - classic feature of AL amyloidosis
DIAGNOSIS
| Method | Finding |
|---|---|
| Biopsy + Congo red stain | Gold standard - apple-green birefringence under polarized light |
| Abdominal fat pad aspirate | Safest, least invasive biopsy site |
| Rectal biopsy | Sensitive, commonly used |
| Bone marrow biopsy | Useful in AL |
| Serum/urine electrophoresis | Monoclonal protein in AL |
| SAA levels | Elevated in AA |
| Immunohistochemistry / Mass spectrometry | Type-specific identification of fibril protein |
| Serum free light chain assay | AL amyloidosis |
| SAP scintigraphy | Extent of organ involvement |
CLINICAL FEATURES SUMMARY
- Renal: nephrotic-range proteinuria, progressive renal failure (commonest manifestation of systemic amyloidosis)
- Cardiac: restrictive cardiomyopathy, arrhythmias, heart failure
- Hepatic: hepatomegaly, rarely significant dysfunction
- GI: malabsorption, motility disorders, bleeding
- Neurologic: peripheral sensorimotor neuropathy, autonomic neuropathy (especially ATTR)
- Other: macroglossia (AL), periorbital purpura (AL - "raccoon eyes"), carpal tunnel syndrome
KEY EXAM POINTS (One-Liners)
- Amyloid = misfolded proteins in β-pleated sheet conformation deposited extracellularly
- Fibril diameter = 8-10 nm, non-branching
- Congo red + polarized light = apple-green birefringence - pathognomonic
- AL type = most common systemic form; associated with plasma cell dyscrasias
- AA type = secondary to chronic inflammation; RA is the most common current association
- SAA is the serum precursor of AA protein; synthesized in liver; induced by IL-6 and IL-1
- Kidney is the most commonly affected organ in systemic amyloidosis
- Sago spleen = follicular deposits; Lardaceous spleen = sinusoidal deposits
- FMF (Familial Mediterranean fever) = most common heredofamilial form; autosomal recessive; AA type
- TTR amyloidosis = familial (mutant TTR) or senile (wild-type TTR); cardiac > neurologic
- Macroglossia = characteristic of AL amyloidosis (plasma cell disorders)
- Normal protein quality control: proteasomes (intracellular) + macrophages (extracellular)
Sources: Robbins, Cotran & Kumar Pathologic Basis of Disease (10th ed.); Robbins & Kumar Basic Pathology
This is a shared conversation. Sign in to Orris to start your own chat.