Hepatitis
Reading File
Asking for Preferences
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Here is a comprehensive overview of the pathophysiology and liver damage mechanisms in hepatitis, drawn from authoritative sources including Robbins, Cotran & Kumar Pathologic Basis of Disease, Harrison's Principles of Internal Medicine 22E, and Sherris & Ryan's Medical Microbiology.
Hepatitis: Pathophysiology & Liver Damage Mechanisms
1. Overview of the Liver's Vulnerability
The liver receives blood from two sources: the portal vein (60-70%) and the hepatic artery (30-40%), making it continuously exposed to gut-derived antigens, toxins, and pathogens. Hepatocytes are organized in zones (zone 1 = periportal, zone 3 = pericentral), and the zone in which injury begins often reflects the nature of the insult. Despite enormous functional reserve, widespread hepatic damage ultimately leads to decompensation.
2. Core Modes of Hepatocyte Death
All forms of hepatitis converge on two fundamental mechanisms of hepatocyte killing:
A. Necrosis
- Ion pump failure disrupts membrane integrity, causing water influx and cell swelling/rupture
- Cytoplasmic constituents (including damage-associated molecular patterns, DAMPs) leak into the extracellular space, alerting innate immune cells
- Macrophages cluster at necrotic foci and phagocytose debris
- Predominant in ischemic/hypoxic injury and oxidative stress
B. Apoptosis
- "Programmed" cell death triggered by cytotoxic T lymphocytes (CTLs) or viral ligands binding death receptors
- Hepatocytes shrink, nuclei condense (pyknosis) then fragment (karyorrhexis), and cells break into eosinophilic acidophil bodies (also called Councilman bodies in yellow fever)
- This is the dominant mechanism in viral hepatitis
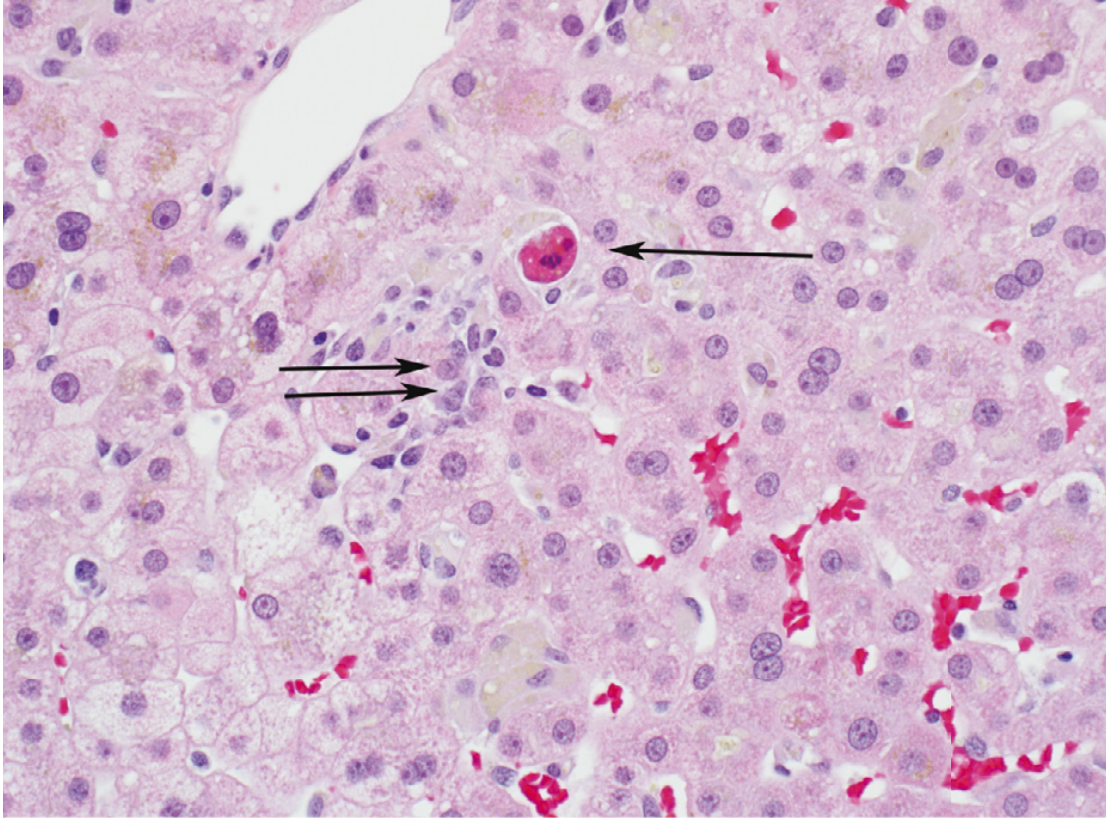
3. Virus-Specific Immunopathology
The key principle: hepatitis viruses are not inherently cytopathic in most cases - the immune response itself causes the liver damage.
Hepatitis A (HAV)
- Acute, self-limited; does not cause chronic infection
- HAV enters via the fecal-oral route, replicates in hepatocytes, and is shed in bile
- Hepatocyte damage is primarily immunopathologic: CD8+ CTLs recognize and destroy infected hepatocytes
- An activated innate immune response (interferons, NK cells) also contributes early
- In most patients, damage resolves completely; fulminant hepatic failure is rare (<1%)
Hepatitis B (HBV)
- The pathogenesis is primarily immune-mediated, not direct viral cytotoxicity
- Acute phase: Circulating immune complexes (HBsAg + antibody) activate complement, causing the serum sickness-like prodrome (rash, arthritis, fever) before jaundice appears. These complexes also deposit in the kidney, causing glomerular damage
- The main pathogenic mechanism: HBV-specific CD8+ CTLs destroy infected hepatocytes. This is protective (clears infection) but simultaneously causes liver damage
- Immune tolerance vs. immune active disease: Neonates infected at birth develop immunologic tolerance to HBV - T cells fail to mount a strong response, so acute liver injury is minimal, but chronic, lifelong infection is nearly universal. In contrast, adults who acquire HBV mount a robust CTL response, suffer acute hepatitis, but clear the virus in ~95% of cases
- Antibody to HBsAg is protective and marks resolution; antibody to HBcAg does NOT protect and marks ongoing viremia
| Age at Infection | Immune Response | Acute Hepatitis | Chronicity |
|---|---|---|---|
| Neonate | Tolerant | Absent/mild | ~90% |
| Adult | Robust | Present | ~5% |
Hepatitis C (HCV)
- HCV is highly efficient at evading host immune responses - this is why it establishes chronic infection in ~75% of cases
- Pattern recognition receptors (PRRs) detect HCV motifs → interferons are produced → activation of innate and adaptive immunity
- Intrahepatic HLA class I-restricted CTLs target nucleocapsid and nonstructural HCV proteins, causing hepatocyte destruction
- However, virus-specific CTL responses are often weak, exhausted, or functionally impaired in chronic HCV, allowing persistent viremia and ongoing low-grade inflammation
- Genetic host factors matter: MHC class II DR5 allele is associated with lower viral replication and better prognosis
- The severity of fibrosis is independent of HCV RNA levels - it is driven by the host inflammatory response, not viral load per se
Hepatitis D (HDV)
- HDV is a defective virus - it requires HBsAg (from HBV) to form its envelope and cannot infect independently
- Liver damage is most likely immune-mediated, with HDV inducing a strong innate immune response
- Co-infection (HBV + HDV simultaneously) typically causes acute, self-limited hepatitis; superinfection (HDV in a chronic HBV carrier) causes a more severe, rapidly progressive disease
4. The Common Pathologic Pathway: From Inflammation to Fibrosis
Regardless of the specific virus, ongoing hepatic injury activates a shared fibrogenic pathway:
Stellate Cell Activation - The Central Driver of Fibrosis
In the normal liver, hepatic stellate cells (located in the space of Disse) are quiescent and store vitamin A (lipid droplets). With chronic injury:
- Activated Kupffer cells and macrophages release TNF-α, PDGF, TGF-β, ET-1, and CCL2
- These signals drive stellate cell transformation into activated myofibroblasts
- Myofibroblasts deposit type I and III collagen, progressively replacing functional liver parenchyma with scar tissue
- PDGF drives stellate cell proliferation and chemotaxis; TGF-β drives fibrosis; ET-1 drives contraction (contributing to portal hypertension)
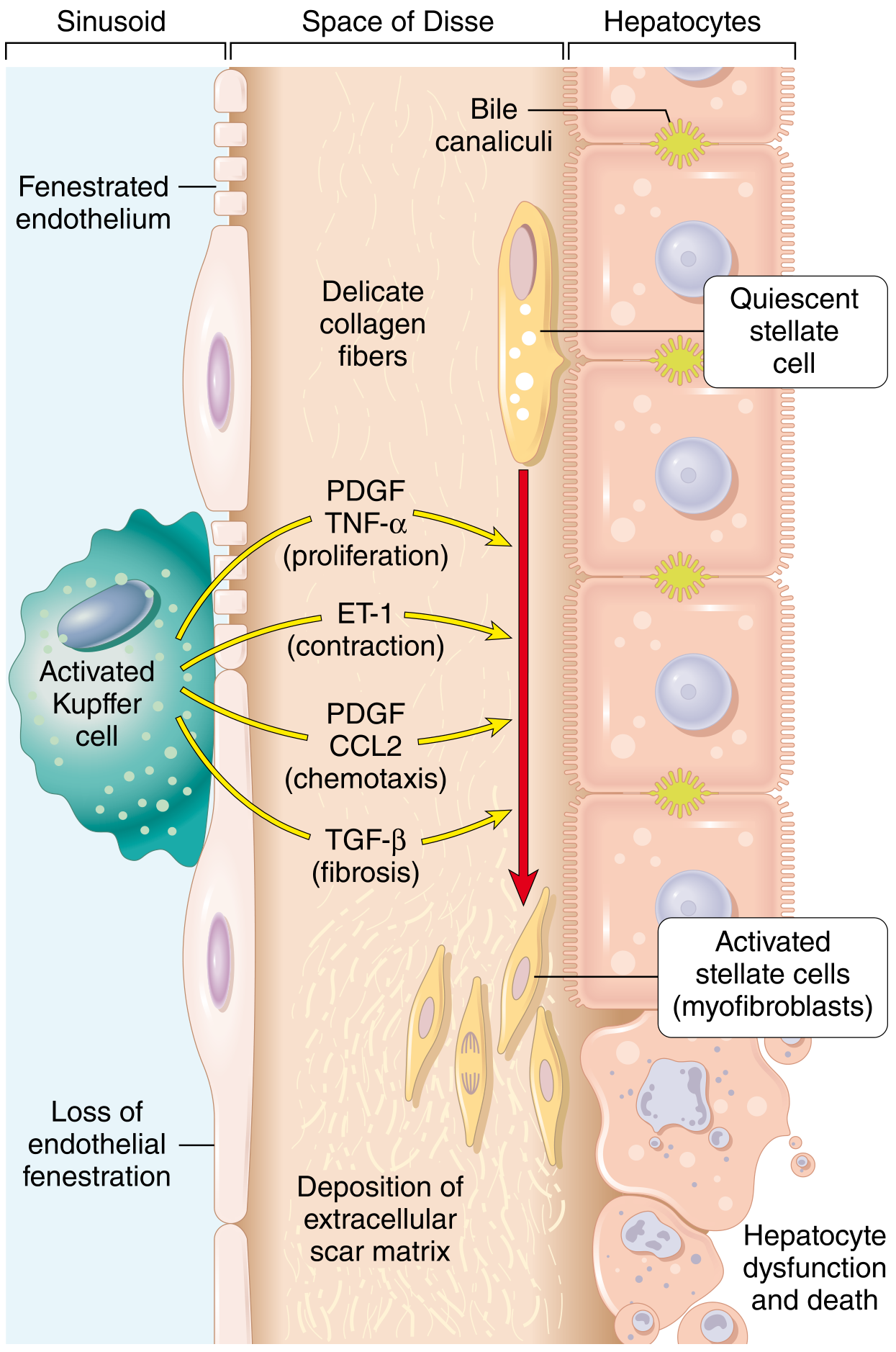
Progression of Necrosis Patterns
- Zone 3 dropout → earliest change (pericentral, most metabolically active, lowest O2)
- Bridging necrosis → necrosis links central vein to portal tract, or portal tract to portal tract; marks a tipping point toward chronicity
- Pan-acinar necrosis → entire lobule destroyed; seen in fulminant hepatic failure
- Confluent necrosis → reticulin framework collapses; the resulting space fills with macrophages and fibrosis
Cirrhosis: The End Result
- In chronic active hepatitis, necrosis + continued inflammation leads to:
- Collapse of hepatic reticulin framework
- Progressive fibrosis (fibrous septa encircle surviving hepatocytes)
- Formation of regenerative nodules (hepatocytes attempting to restore parenchyma)
- Architectural distortion → portal hypertension, synthetic dysfunction
- If the injuring stimulus is removed (e.g., viral clearance, alcohol cessation), stellate cell activation ceases and metalloproteinases produced by hepatocytes can partially break down fibrous septa ("incomplete septal cirrhosis"), but vascular remodeling often does not fully reverse, explaining persistent portal hypertension even after viral cure
5. Immune-Mediated Mechanisms Summary
| Mechanism | Hepatitis A | Hepatitis B | Hepatitis C | Hepatitis D |
|---|---|---|---|---|
| CD8+ CTL killing | Yes | Yes (primary) | Yes (primary) | Indirect |
| Immune complex/complement | Minor | Yes (prodrome) | Minor | Minor |
| Innate immune (IFN, NK) | Yes | Yes | Yes (evaded by HCV) | Strong |
| Tolerance/persistence | No | Yes (neonates) | Yes (adults) | Via HBV |
6. Acute Liver Failure
When >80-90% of hepatic function is lost, liver failure ensues. In the context of viral hepatitis:
- Hepatocyte necrosis is a combination of direct viral toxicity + immune-mediated destruction
- The mechanism differs from acetaminophen toxicity (which is purely toxic/metabolic, occurring within days) - viral hepatitis-induced failure develops over weeks to months
- Massive hepatic necrosis leaves islands of preserved or regenerating hepatocytes surrounded by broad areas of parenchymal loss; the liver shrinks in size
- Without transplantation, mortality is ~80%
Key sources:
- Robbins, Cotran & Kumar - Pathologic Basis of Disease (Mechanisms of Injury, Stellate Cell Activation, Acute Liver Failure sections)
- Harrison's Principles of Internal Medicine, 22E (HBV immune tolerance/immune active phases, HCV pathogenesis)
- Sherris & Ryan's Medical Microbiology, 8th Ed. (Virus-induced immunopathology, HBV and HCV pathogenesis)
This is a shared conversation. Sign in to Orris to start your own chat.