Intertesial lung disease
Reading File
Finding Sources
Searching PubMed
"interstitial lung disease" diagnosis management
Reading File
Finding Sources
Reading File
Reading File
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Reading File
Finding Sources
Reading File
Loading Image
Loading Image
Here is a comprehensive overview of Interstitial Lung Disease (ILD), drawn from Murray & Nadel's Textbook of Respiratory Medicine and supplemented with current evidence.
Interstitial Lung Disease (ILD)
Definition
Interstitial lung disease (ILD) refers to a heterogeneous group of diffuse parenchymal infiltrative disorders that impair gas exchange by disrupting the alveolar walls. In many ILDs, lung regeneration fails and interstitial fibrosis - from fibroblastic proliferation and excessive collagen deposition - becomes the dominant pathologic process. Nonfibrotic processes can also cause ILD through abnormal infiltration by cells (eosinophils, histiocytes) or substances (amyloid).
Note: The term "interstitial" is somewhat misleading - pathologic involvement is not restricted to the anatomic interstitium (the space between the basement membranes of alveolar epithelium and capillary). Many ILDs involve the alveolar spaces as well.
Epidemiology
- Prevalence: ~81/100,000 in men, ~67/100,000 in women
- Incidence: ~32/100,000/year in men, ~26/100,000/year in women
- Increases markedly with age, especially over 75
- US mortality from ILD doubled from 1980 to 2014
Classification
ILDs are most practically classified by whether a cause is identifiable:
| Known Cause | Unknown Cause |
|---|---|
| Connective tissue disease-related ILDs | Idiopathic pulmonary fibrosis (IPF) and other idiopathic interstitial pneumonias (IIPs) |
| Environmental and occupational ILDs | Sarcoidosis |
| Drug- and radiation-related ILDs | Idiopathic eosinophilic pneumonias |
| Smoking-related ILDs | Vasculitis |
| Rare forms (LAM, pulmonary alveolar proteinosis, amyloidosis, alveolar microlithiasis) | Rare forms (IgG4-related disease, Erdheim-Chester disease) |
- Murray & Nadel's Textbook of Respiratory Medicine, Table 89.1
Idiopathic Interstitial Pneumonias (IIPs) - Key Subtypes
The ATS/ERS classification recognizes the following IIPs:
| Pattern | Clinical Entity |
|---|---|
| Usual interstitial pneumonia (UIP) | IPF (most common IIP, 20-30% of all ILDs) |
| Nonspecific interstitial pneumonia (NSIP) | NSIP (often CTD-associated) |
| Respiratory bronchiolitis | RB-ILD |
| Desquamative interstitial pneumonia | DIP |
| Cryptogenic organizing pneumonia | COP |
| Acute interstitial pneumonia | AIP (Hamman-Rich syndrome) |
| Lymphoid interstitial pneumonia | LIP |
Focus: Idiopathic Pulmonary Fibrosis (IPF)
IPF is the most common and most studied ILD. It is a progressive parenchymal fibrosis of unknown cause.
Histopathology
The hallmark is the Usual Interstitial Pneumonia (UIP) pattern:
- Heterogeneous, subpleural and paraseptal distribution
- Areas of normal lung interspersed with dense fibrosis ("temporal heterogeneity")
- Microscopic honeycombing - enlarged air spaces lined by bronchiolar epithelium, filled with mucin
- Fibroblastic foci at the edges of scarred areas
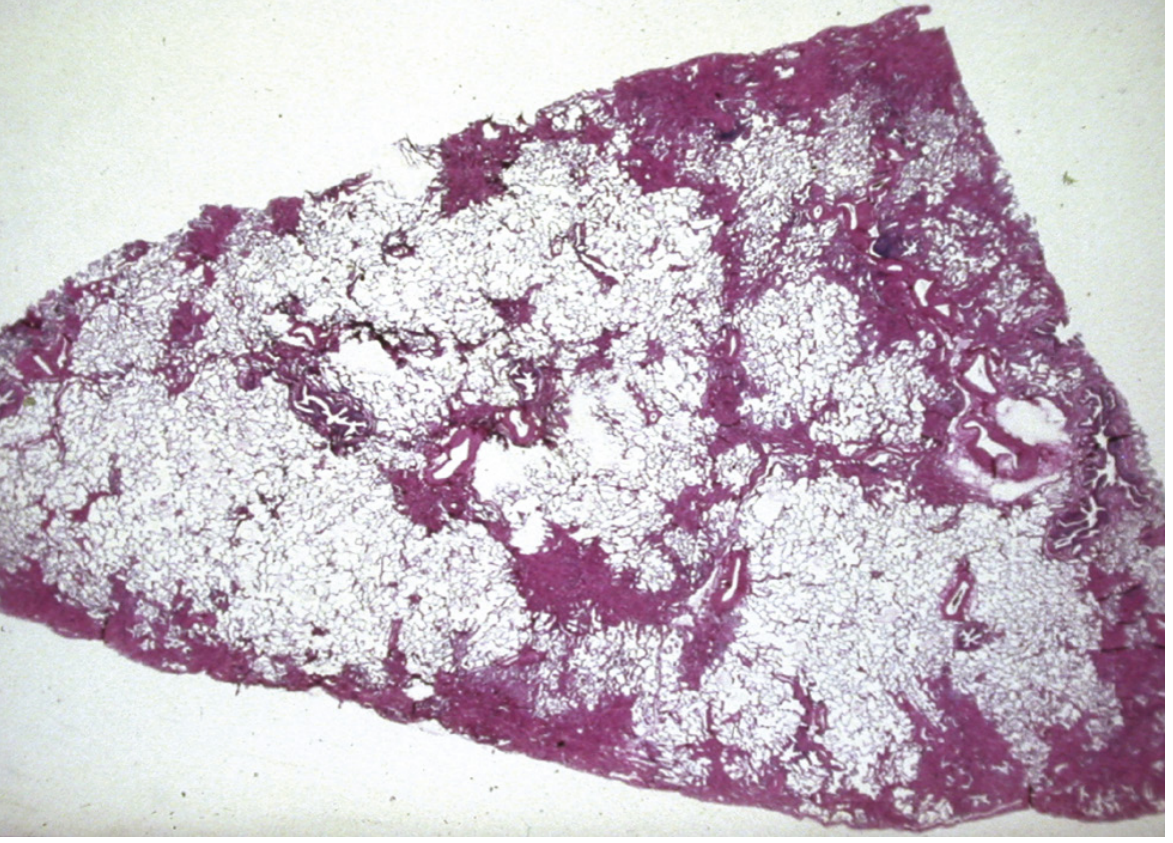
Clinical Features
- Age usually >60 years
- Progressive exertional dyspnea (gradual onset over months to years)
- Dry, persistent cough
- Velcro-like inspiratory crackles at lung bases (bibasal)
- Digital clubbing in ~50%
- Late-stage: cyanosis, cor pulmonale, right heart failure
Chest Imaging
Chest X-ray:
- Peripheral reticular opacities, netlike appearance
- Lower zone predominance
- Reduced lung volumes
- Honeycombing in advanced disease
HRCT (diagnostic cornerstone):
- Peripheral (subpleural) and basal predominant opacities
- Reticulation with or without traction bronchiectasis
- Honeycombing (3-5mm cysts sharing walls, stacking in rows)
- IPF can be diagnosed without biopsy if HRCT shows typical/definite or probable UIP pattern in the right clinical context
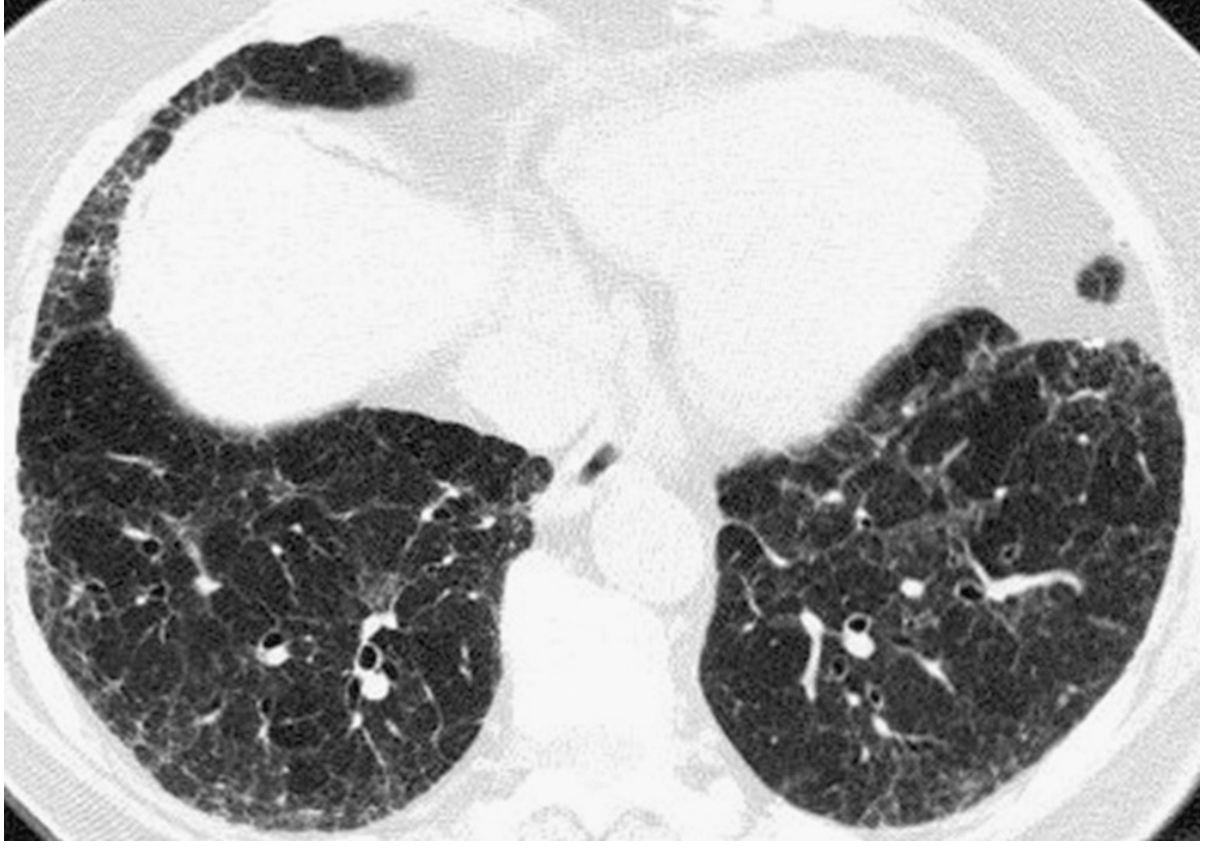
Pulmonary Function Tests
- Restrictive pattern: reduced TLC, FVC, RV
- Reduced DLCO (diffusing capacity) - due to loss of pulmonary capillary volume and V/Q mismatch
- Resting hypoxemia + respiratory alkalosis
- Exercise worsens A-a O2 gradient
- 6-minute walk distance (6MWD) is a strong predictor of mortality
Diagnosis
Diagnosis is established by:
- HRCT showing typical or probable UIP pattern PLUS
- Appropriate clinical context (age >60, no significant inhalational/drug exposure, no connective tissue disease evidence)
If HRCT is indeterminate, surgical lung biopsy (VATS) may be needed. Transbronchial cryobiopsy is an emerging option. Molecular classifiers (genomic classifiers from BAL or biopsy) are increasingly available.
The UIP pattern on HRCT or pathology can also be seen in chronic hypersensitivity pneumonitis, CTD-ILD, and drug-induced ILD - so clinical context is essential.
Approach to Suspected ILD - General Workup
| Step | Details |
|---|---|
| History | Duration, occupational/drug/inhalant exposure, systemic symptoms, GER (aspiration), family history |
| Physical exam | Bibasal crackles, clubbing, extrapulmonary signs of CTD |
| Chest HRCT | Best initial imaging - characterizes pattern and distribution |
| PFTs | Spirometry, lung volumes, DLCO |
| Labs | ANA, RF, anti-CCP, Scl-70, Jo-1, MPO/PR3 ANCA, HP panel (precipitins/IgG) |
| BAL | Differential cell count - lymphocytosis (HP, NSIP, sarcoid), eosinophilia, neutrophilia |
| Bronchoscopy | BAL + transbronchial biopsy (for granulomatous diseases, infections) |
| Lung biopsy | VATS surgical biopsy when diagnosis uncertain |
Treatment
IPF-Specific Antifibrotics
Two FDA-approved antifibrotic drugs slow progression in IPF:
| Drug | Mechanism | Effect |
|---|---|---|
| Nintedanib (Ofev) | Pan-tyrosine kinase inhibitor (PDGFR, VEGFR, FGFR) | Reduces annual FVC decline by ~50% |
| Pirfenidone (Esbriet) | Antifibrotic, anti-inflammatory | Reduces FVC decline and mortality |
Nintedanib has also been shown to slow FVC decline in SSc-associated ILD and in progressive fibrosing ILDs beyond IPF (SENSCIS and INBUILD trials).
Supportive & Comorbidity Management
- Supplemental oxygen for resting or exertional hypoxemia
- Pulmonary rehabilitation
- Proton pump inhibitors (GER/aspiration is a potential trigger)
- Pulmonary hypertension management (present in up to 44% of advanced IPF)
- Lung transplantation - the only potentially curative option; considered for eligible patients with progressive disease
Acute Exacerbations of IPF
- Sudden acceleration of respiratory decline
- New bilateral ground-glass opacities superimposed on UIP pattern
- High mortality (50-90%)
- Treated with high-dose corticosteroids, broad-spectrum antibiotics
Prognosis of IPF
- Median survival after diagnosis: ~3 years
- 5-year survival: only 20-35%
- IPF itself (from acute exacerbation or gradual progression) is the cause of death in ~50% of cases
- Cardiovascular disease accounts for ~20% of deaths
- Pulmonary hypertension found in 45% at autopsy
- Adverse prognostic factors: older age, male sex, lower FVC, lower DLCO, honeycombing on HRCT, shorter 6MWD, higher mPAP
Other Key ILDs at a Glance
| ILD | Key Features |
|---|---|
| Hypersensitivity Pneumonitis (HP) | Exposure to organic antigens (birds, molds, hot tubs); upper/mid-zone fibrosis; lymphocytosis on BAL |
| Sarcoidosis | Upper lobe peribronchovascular nodules; bilateral hilar adenopathy; non-caseating granulomas |
| CTD-ILD | NSIP pattern common; associated with RA, SSc, polymyositis/DM, SLE, Sjogren's |
| Drug-induced ILD | Methotrexate, amiodarone, bleomycin, nitrofurantoin; variable patterns |
| Asbestosis | Occupational exposure; basal fibrosis + pleural plaques |
| COP | Peripheral airspace consolidation; responds well to steroids |
Recent Evidence (2024-2026)
- A 2025 systematic review (PMID 40876558) confirmed nintedanib and tocilizumab as key treatments for RA-associated ILD
- A 2025 meta-analysis (PMID 39083028) reviewed anti-synthetase syndrome ILD management, underscoring the role of immunosuppressants
- ERS 2024 guidelines (PMID 39147411) were issued for pulmonary alveolar proteinosis, a rare ILD
Sources: Murray & Nadel's Textbook of Respiratory Medicine, 2-Volume Set - Chapters 89, 90, 91; PubMed PMID 40876558, 39083028, 39147411
This is a shared conversation. Sign in to Orris to start your own chat.