Von willi brand disease
Reading File
Finding Sources
Searching PubMed
"von Willebrand disease"[MeSH Terms]
Reading File
Reading File
Reading File
Reading File
Loading Image
I now have comprehensive information from multiple authoritative sources. Here is a complete overview of von Willebrand Disease (vWD):
Von Willebrand Disease (vWD)
Overview
Von Willebrand disease is the most common inherited bleeding disorder, affecting approximately 1% of the population. It is caused by quantitative or qualitative deficiencies in von Willebrand factor (vWF), a large multimeric plasma glycoprotein with two essential roles in hemostasis. Clinically significant bleeding symptoms occur in roughly 1 in 1,000 individuals. — Goldman-Cecil Medicine
Pathophysiology
The VWF gene has 52 exons spanning 178 kb on chromosome 12. vWF is produced exclusively by endothelial cells and megakaryocytes.
- In endothelial cells, vWF is stored in Weibel-Palade bodies and released by secretagogues such as histamine, thrombin, and desmopressin (DDAVP)
- In platelets, vWF is stored in α-granules and accounts for 10–20% of total vWF in platelet-rich plasma
vWF serves two critical hemostatic roles:
- Platelet adhesion — binds exposed subendothelial collagen (A3 domain → types I/III; A1 domain → types IV/VI) and platelets via GpIb receptor
- Factor VIII carrier — circulates with factor VIII in a noncovalent complex, protecting it from proteolytic degradation
High-molecular-weight (HMW) multimers are the most functionally active form. The metalloprotease ADAMTS13 cleaves vWF multimers to regulate their size in plasma. Normal plasma vWF: 50–150 IU/dL.
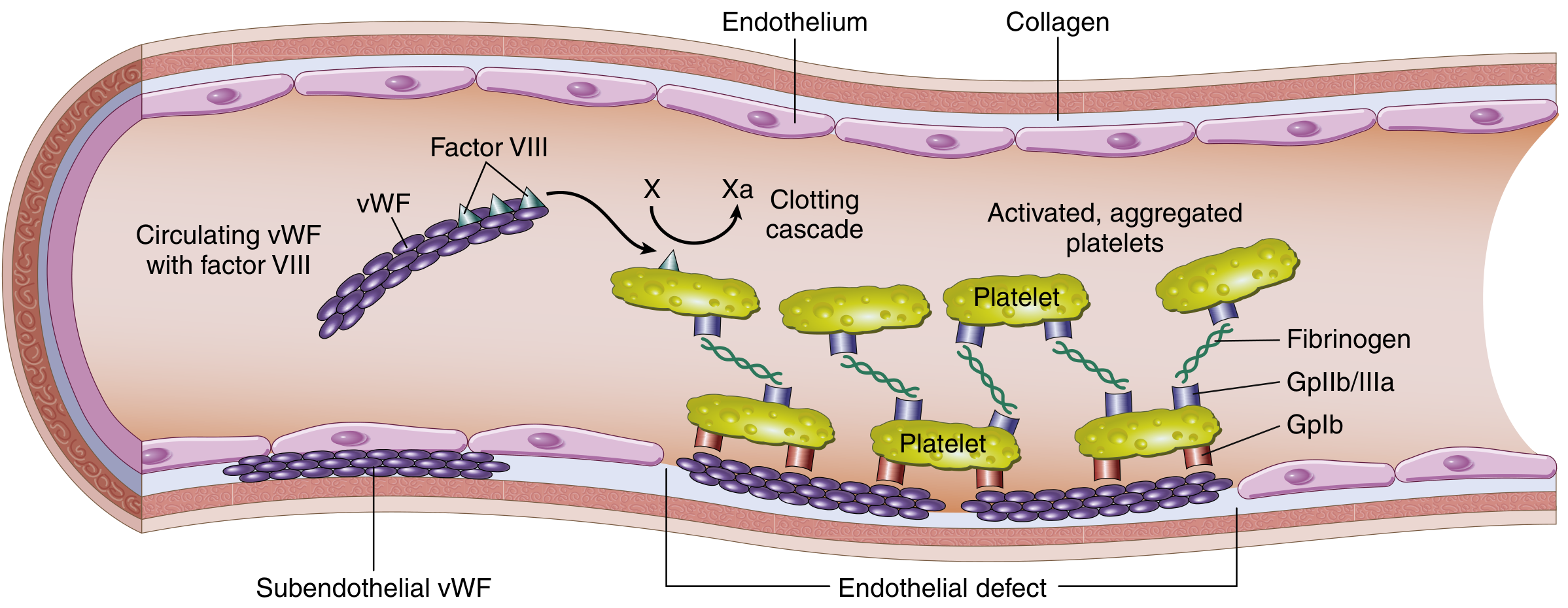
Structure and function of the Factor VIII–vWF complex. vWF circulates with Factor VIII, adheres to subendothelial collagen, and bridges platelets via GpIb, driving platelet plug formation. — Robbins & Kumar Basic Pathology
Classification
| Type | Mechanism | Inheritance | Frequency |
|---|---|---|---|
| Type 1 | Partial quantitative reduction in vWF (< 50 IU/dL) | Autosomal dominant | ~75% of cases (most common) |
| Type 2A | Loss of HMW multimers — not synthesized | AD | ~10–15% |
| Type 2B | Abnormal "hyperfunctional" HMW multimers rapidly cleared; causes spontaneous platelet aggregation and mild thrombocytopenia | AD | Rare |
| Type 2M | Defective platelet binding despite normal multimer distribution | AD | Rare |
| Type 2N | Defective Factor VIII binding — mimics hemophilia A | AR | Rare |
| Type 3 | Complete absence of vWF; compound/homozygous; severe | AR | 1–5% |
Clinical Features
vWD produces compound defects in platelet function and coagulation, but in most patients only the platelet defect is clinically apparent:
- Mucocutaneous bleeding: epistaxis, gingival bleeding, easy bruising
- Menorrhagia (a major presenting symptom in females)
- Gastrointestinal bleeding
- Excessive bleeding from wounds or surgery
- Hemarthroses are rare (unlike hemophilia)
- Factor VIII activity is typically in the 6–50% range in most subtypes
- Type 3 and rare homozygous patients can develop severe Factor VIII deficiency resembling hemophilia A
Laboratory Findings
| Test | Typical Finding |
|---|---|
| Bleeding time / PFA-100 | Prolonged |
| aPTT | Slightly prolonged (if Factor VIII low) |
| PT | Normal |
| Platelet count | Normal (except mildly low in type 2B) |
| vWF antigen | Reduced (type 1, 3) or normal (type 2) |
| vWF ristocetin cofactor activity | Reduced |
| Factor VIII activity | Normal or mildly reduced |
| Multimer analysis | Absent HMW multimers in type 2A/2B |
Treatment
Mild to moderate vWD:
- Desmopressin (DDAVP) 0.3 mcg/kg IV — triggers release of vWF from Weibel-Palade bodies; preferred first-line due to low risk and low cost
- Tranexamic acid (antifibrinolytic) — effective adjunct, especially for mucosal bleeding and menorrhagia
Severe vWD or DDAVP-unresponsive:
- Factor VIII/vWF concentrate (lyophilized) at 50 IU/kg — replacement therapy
- A unique feature: transfusion of plasma components triggers a progressive rise in Factor VIII lasting 12–40 hours, so subsequent doses can be smaller and less frequent
- Fresh frozen plasma or cryoprecipitate in emergencies
Acquired von Willebrand Syndrome
A non-hereditary form occurring without prior personal/family history:
- Associated conditions: lymphoproliferative disorders (MGUS, myeloma, Waldenström, NHL, CLL), myeloproliferative disorders, autoimmune disease, aortic stenosis, ECMO, LVADs
- Mechanisms: anti-vWF antibodies causing rapid clearance; high-shear states causing enhanced ADAMTS13 proteolysis of HMW multimers
- Treatment: treat underlying condition; options include DDAVP, vWF concentrate, IVIg (especially for paraprotein-associated), tranexamic acid, plasmapheresis, recombinant Factor VIIa
Key Distinctions from Hemophilia
| Feature | vWD | Hemophilia A |
|---|---|---|
| Inheritance | Autosomal dominant (types 1, 2) | X-linked recessive |
| Sex | Males and females equally | Primarily males |
| Bleeding pattern | Mucocutaneous | Deep (hemarthroses, muscle) |
| vWF | Low or dysfunctional | Normal |
| Factor VIII | Normal or mildly low | Severely low |
Sources: Robbins & Kumar Basic Pathology p. 430 | Goldman-Cecil Medicine Ch. 159 | Rosen's Emergency Medicine Ch. 111
This is a shared conversation. Sign in to Orris to start your own chat.