Mucopolysachharides
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Reading File
Reading File
Reading File
Finding Sources
I now have comprehensive coverage from multiple authoritative sources. Let me compile the full answer.
Mucopolysaccharides (Glycosaminoglycans) and the Mucopolysaccharidoses (MPS)
What Are Mucopolysaccharides?
Mucopolysaccharides - now more correctly called glycosaminoglycans (GAGs) - are long-chain, sulfated polysaccharides that form the carbohydrate side-chains of proteoglycans found in connective tissues, bone, cartilage, and extracellular matrix. The major GAGs include:
- Dermatan sulfate
- Heparan sulfate
- Keratan sulfate
- Chondroitin sulfate (4- and 6-sulfate)
- Hyaluronan (not sulfated)
They are normally degraded within lysosomes by a series of specific hydrolases. When these enzymes are deficient, GAGs accumulate within lysosomes throughout the body - in the brain, spinal cord, heart, viscera, bone, and connective tissue - producing a group of diseases called the Mucopolysaccharidoses (MPS).
The overall prevalence of MPS is approximately 1 in 8,000 births.
General Features of All MPS Disorders
- Storage of GAGs within lysosomes (lysosomal storage diseases)
- Progressive accumulation due to defective degradation
- Multisystem involvement: skeletal, cardiac, respiratory, neurologic, ocular, hepatosplenic
- All are autosomal recessive except MPS II (Hunter), which is X-linked recessive
- Diagnosis: urinary GAG excretion pattern + specific enzyme assay + gene analysis
- Characteristic "gargoyle" or coarse facial features in several types
Classification of the Mucopolysaccharidoses
| Type | Eponym | Enzyme Deficiency | GAGs Excreted | Key Features |
|---|---|---|---|---|
| MPS I H | Hurler syndrome | α-L-Iduronidase | Dermatan sulfate, Heparan sulfate | Most severe; corneal clouding, coarse facies, intellectual disability, organomegaly, kyphosis, cardiac/respiratory death by mid-adolescence |
| MPS I S | Scheie syndrome | α-L-Iduronidase (partial) | Dermatan sulfate, Heparan sulfate | Milder allele; normal intelligence, normal life span |
| MPS I H/S | Hurler-Scheie | α-L-Iduronidase (intermediate) | Dermatan sulfate, Heparan sulfate | Severe somatic disease, usually without major neurologic degeneration |
| MPS II | Hunter syndrome | Iduronate sulfate sulfatase | Dermatan sulfate, Heparan sulfate | X-linked; similar to Hurler but milder; no corneal clouding |
| MPS III A-D | Sanfilippo syndrome | 4 different enzymes (see below) | Heparan sulfate | Most common MPS; predominant CNS involvement (behavioral, intellectual decline, seizures); mild somatic features |
| MPS IV A/B | Morquio syndrome | Galactosamine-6-sulfatase (A) / β-Galactosidase (B) | Keratan sulfate, Chondroitin 6-sulfate | Normal intelligence; prominent skeletal dysplasia, kyphoscoliosis, odontoid hypoplasia, cervical instability risk |
| MPS V | (No longer used) | - | - | Reclassified as Scheie (now MPS I S) |
| MPS VI | Maroteaux-Lamy | N-acetylgalactosamine-4-sulfatase (Arylsulfatase B) | Dermatan sulfate | Hurler-like somatic features; normal intelligence; corneal clouding; cardiac valve disease |
| MPS VII | Sly syndrome | β-Glucuronidase | Dermatan sulfate, Heparan sulfate, Chondroitin 4-sulfate | Wide severity range; can present as fetal hydrops |
Sources: Adams and Victor's Principles of Neurology 12th Ed; Harrison's Principles of Internal Medicine 22E (2025); Emery's Elements of Medical Genetics and Genomics
Individual Syndromes - Clinical Detail
MPS I - Hurler Syndrome
- Onset: end of the first year of life
- Clinical triad: coarse "gargoyle" facies, corneal clouding, intellectual disability
- Skeletal: dwarfism, kyphosis, broad hands with stubby fingers, flexion contractures at knees and elbows
- Hearing loss (conductive), hepatosplenomegaly, valvular heart disease, recurrent respiratory infections, hernias
- Biochemistry: absence of α-L-iduronidase → accumulation of dermatan + heparan sulfate in tissues and urine
- Also increased ganglioside content in brain neurons
- Death: mid-adolescence from cardiac failure and respiratory infections
- Diagnosis: metachromatic granules in leukocytes; increased urinary GAGs; confirmed by enzyme assay (α-L-iduronidase) and IDUA gene analysis
MPS II - Hunter Syndrome
- X-linked recessive (only X-linked MPS)
- Onset: 2-5 years; males affected
- Hurler-like but milder: coarse facies, joint stiffness, deafness, organomegaly, developmental delay
- No corneal clouding (key distinguishing feature from Hurler)
- Two forms: severe (death in mid-teens) and mild (relatively normal intelligence, survival to middle age)
- Enzyme deficiency: iduronate sulfate sulfatase (IDS gene)
- Urine: excess dermatan + heparan sulfate
MPS III - Sanfilippo Syndrome
- Most common MPS
- Predominant neuropsychiatric presentation: behavioral problems, intellectual regression, hyperactivity, seizures
- Mild somatic features (unlike other MPS types)
- Four subtypes (A-D) from four different enzymes, all degrading heparan sulfate:
- IIIA: N-sulphoglucosamine sulphohydrolase (SGSH gene)
- IIIB: α-N-acetylglucosaminidase (NAGLU gene)
- IIIC: Heparan-α-glucosaminide N-acetyltransferase (HGSNAT gene)
- IIID: N-acetylglucosamine-6-sulfatase (GNS gene)
- Types A and B account for 90% of cases
- Death: early adult life
MPS IV - Morquio Syndrome
- Onset: 2-3 years
- Skeletal dysplasia without intellectual impairment (intelligence normal)
- Short stature, thoracic deformity, kyphoscoliosis
- Risk: spinal cord compression from odontoid hypoplasia and cervical instability
- Slight corneal clouding; cardiac and respiratory complications
- Urine: keratan sulfate
- Enzyme: galactosamine-6-sulfatase (MPS-IVA, GALNS gene) or β-galactosidase (MPS-IVB, GLB1 gene)
- Prevalence: ~1 in 200,000-300,000
MPS VI - Maroteaux-Lamy Syndrome
- Hurler-like somatic features: coarse facies, short stature, kyphosis, joint restriction, corneal clouding, cardiac valve abnormalities
- Intelligence normal
- Enzyme deficiency: N-acetylgalactosamine-4-sulfatase / arylsulfatase B (ARSB)
- Urine: dermatan sulfate
- Spinal cord compression can occur
- Two forms: severe (survival to early adulthood) and mild
MPS VII - Sly Syndrome
- Enzyme deficiency: β-Glucuronidase (GUSB gene)
- Wide range of severity
- Severe form: fetal hydrops - can cause stillbirth or perinatal death
- Later-onset form: short stature, coarse facies, hepatosplenomegaly
Diagnosis
- Urine GAG quantification and fractionation - screening test (increased dermatan, heparan, keratan, or chondroitin sulfate excretion patterns are type-specific)
- Enzyme activity assay - in serum, leukocytes, or cultured fibroblasts - confirmatory
- Molecular gene analysis - IDUA, IDS, NAGLU, GALNS, GUSB, etc.
- Newborn screening (NBS): MPS I added to recommended NBS panels in several US states; Pompe disease similarly screened
Treatment
Enzyme Replacement Therapy (ERT)
Available FDA-approved ERTs:
- MPS I: Laronidase (recombinant α-L-iduronidase) - effective for visceral/cardiac/respiratory manifestations; does not cross blood-brain barrier (no direct CNS benefit)
- MPS II: Idursulfase - weekly IV; addresses visceral manifestations; no CNS penetration
- MPS IVA: Elosulfase alfa - approved; improves ambulatory mobility and decreases pain; no treatment for MPS IVB
- MPS VI: Galsulfase (arylsulfatase B) - FDA approved
- MPS VII: Vestronidase alfa-vjbk - approved
Hematopoietic Stem Cell Transplantation (HSCT)
- Standard treatment for MPS I presenting at <2 years of age with neurologic involvement
- Stabilizes CNS disease, reverses hepatosplenomegaly, improves cardiac/respiratory disease
- Does not improve corneal disease or progressive skeletal disease
- HSCT has not been successful in treating CNS disease in MPS II
- Not helpful in Hunter or Sanfilippo diseases
- Combination ERT + HSCT is being tried (ERT before transplant to reduce disease burden)
Supportive Management
- For MPS (airway): adenotonsillectomy, CPAP, or tracheotomy (tonsil/adenoid/tongue hypertrophy and tracheobronchial deposits cause obstructive sleep apnea and chronic pulmonary disease - a major cause of death)
- Regular lifelong cardiac and respiratory follow-up even after successful HSCT
Wound Healing Role
Mucopolysaccharides (particularly hyaluronic acid and chondroitin sulfate) also play important roles in wound healing - they facilitate fibroblast migration, cell proliferation, and tissue remodeling during the repair process.
Key Memory Aids:
- "Hunter = Heparan + dermatan + Hemizygous (X-linked)" - the only X-linked MPS
- "Sanfilippo = Smart (normal IQ early) then degenerates + Sulfate of heparan only"
- "Morquio = Musculoskeletal (skeletal dysplasia) + Mind intact (normal intelligence)"
- "Maroteaux-Lamy = like Hurler but Lucid (normal intelligence)"
Sources: Harrison's Principles of Internal Medicine 22E (2025, McGraw Hill); Adams and Victor's Principles of Neurology 12th Ed; Emery's Elements of Medical Genetics and Genomics; Cummings Otolaryngology; Goldman-Cecil Medicine
Explain glucose transporter glut in intestine.
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Excellent - both diagrams retrieved. I now have everything needed for a thorough explanation.
Glucose Transporters (GLUT/SGLT) in the Intestine
Overview
Dietary carbohydrates are digested to monosaccharides (glucose, galactose, fructose) at the brush border by enzymes like lactase, sucrase-isomaltase, and maltase before absorption. Absorption occurs predominantly in the jejunum via three distinct membrane carrier systems:
- SGLT1 (apical/brush-border membrane) - for glucose and galactose
- GLUT5 (apical/brush-border membrane) - for fructose
- GLUT2 (basolateral membrane) - exit route for all three monosaccharides into portal blood
The Classic Diagram (Normal/Low Glucose Load)
(Guyton and Hall Textbook of Medical Physiology)
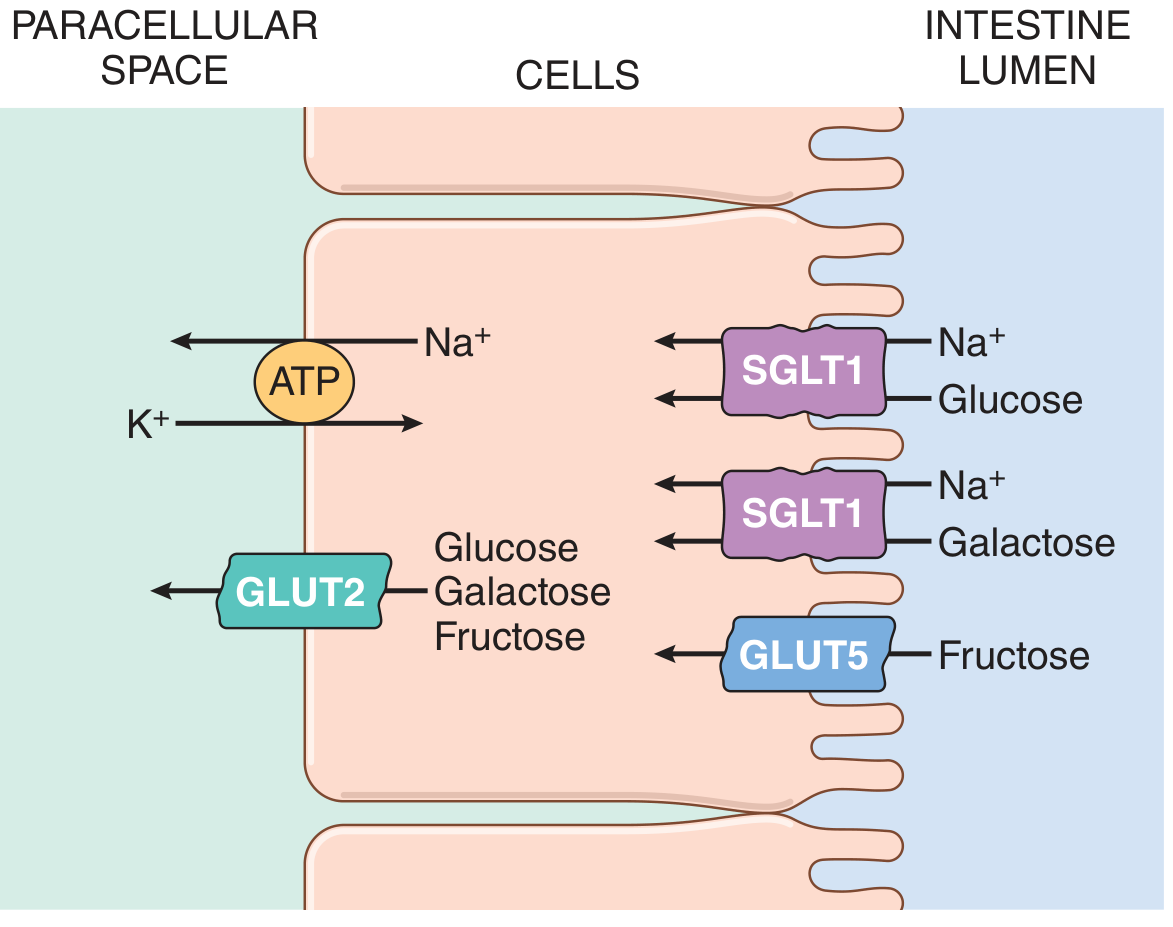
Step-by-Step Mechanism
Step 1: Na⁺/K⁺-ATPase Creates the Driving Force (Basolateral Membrane)
- The Na⁺/K⁺-ATPase pump on the basolateral membrane (BLM) continuously pumps Na⁺ out of the enterocyte and K⁺ in, using ATP
- This keeps intracellular Na⁺ low and maintains an inside-negative membrane potential
- The resulting electrochemical Na⁺ gradient is the ultimate energy source for glucose absorption - this is secondary active transport
Step 2: SGLT1 - Apical/Brush-Border Membrane Entry (Glucose & Galactose)
| Property | Detail |
|---|---|
| Full name | Sodium-Glucose Cotransporter 1 |
| Gene | SLC5A1 |
| Location | Apical/brush-border membrane (BBM) of enterocytes |
| Substrates | Glucose AND galactose (not both simultaneously) |
| Coupling ratio | 2 Na⁺ : 1 monosaccharide per transport cycle |
| Transport type | Secondary active transport (electrogenic) |
| Affinity | High affinity (efficient at low luminal sugar concentrations) |
Mechanism: Na⁺ flows down its electrochemical gradient into the cell through SGLT1, dragging glucose (or galactose) along with it. Since 2 positive charges enter per cycle, the process is electrogenic - it depolarizes the BBM. SGLT1 is a high-affinity transporter, meaning it works well at normal/low luminal glucose concentrations.
Step 3: GLUT5 - Apical Membrane Entry (Fructose Only)
| Property | Detail |
|---|---|
| Full name | Glucose Transporter 5 |
| Gene | SLC2A5 |
| Location | Apical/brush-border membrane |
| Substrate | Fructose only (not glucose or galactose) |
| Transport type | Facilitated diffusion (passive, energy-independent) |
| Na⁺ dependence | None |
- Fructose enters the enterocyte purely by concentration gradient via GLUT5
- No Na⁺, no ATP required
- Rate of fructose absorption is ~half that of glucose/galactose as a result
Step 4: GLUT2 - Basolateral Membrane Exit (All Three Sugars)
| Property | Detail |
|---|---|
| Full name | Glucose Transporter 2 |
| Gene | SLC2A2 |
| Location | Basolateral membrane (under normal conditions) |
| Substrates | Glucose, galactose, and fructose |
| Transport type | Facilitated diffusion (passive) |
| Affinity | Low affinity |
- All three monosaccharides that enter the enterocyte exit across the BLM via GLUT2 into the paracellular space and then into portal blood
- The low affinity is physiologically important - it ensures sugars only exit when intracellular concentrations exceed portal blood concentrations (i.e., net export only when the cell is loaded)
- GLUT2 also functions in the pancreas (β-cells) as a glucose sensor to trigger insulin secretion proportional to blood glucose
The Dynamic Model: GLUT2 Trafficking Under High-Glucose Load
(Sleisenger and Fordtran's Gastrointestinal and Liver Disease, Fig. 102.5)
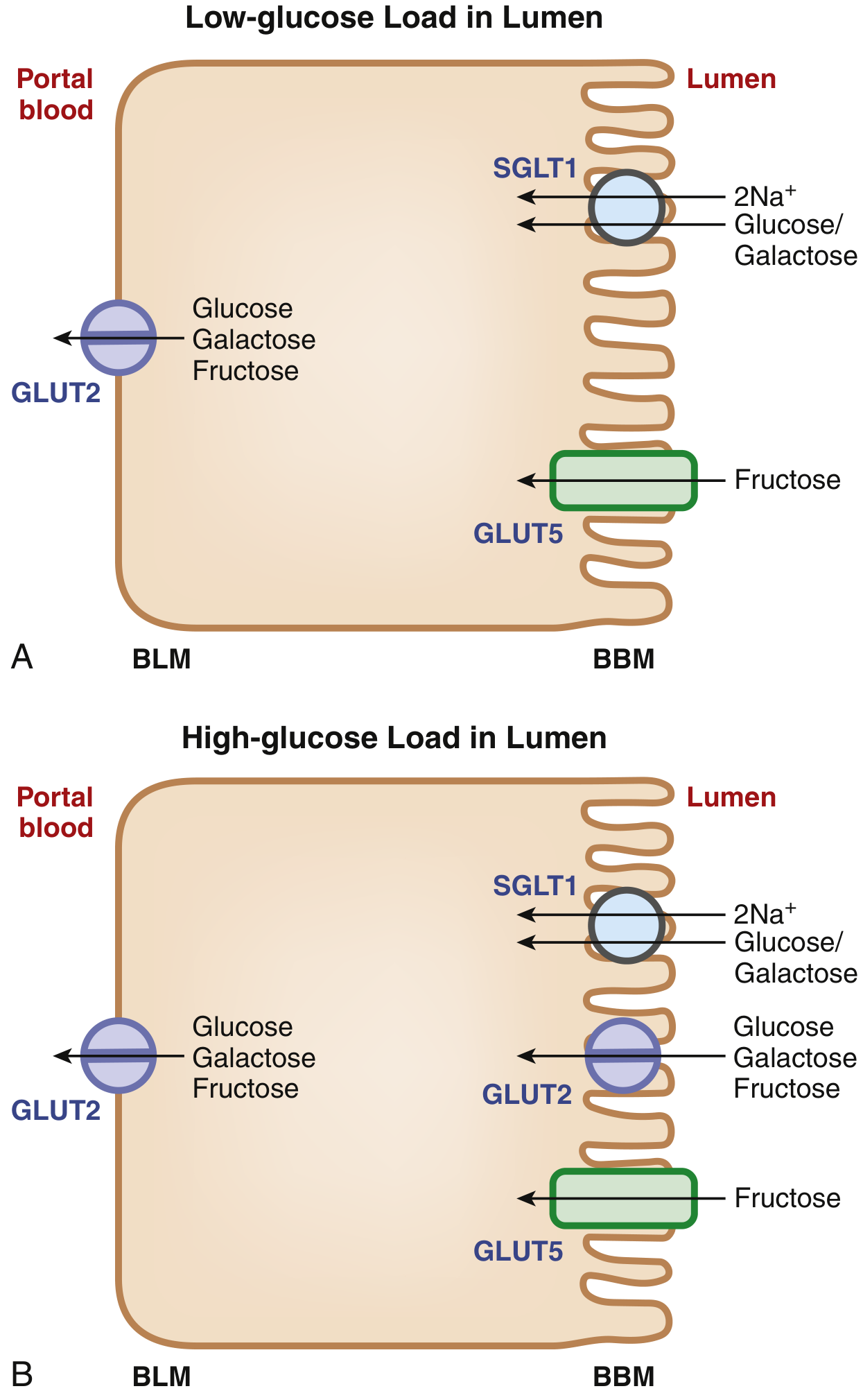
The classical model shows GLUT2 only on the BLM. However, when the intestinal lumen faces a high sugar load:
- SGLT1-mediated glucose entry acts as the signal that triggers trafficking of GLUT2 from the BLM to the BBM (apical membrane)
- GLUT2 then appears on both membranes simultaneously
- Because GLUT2 is a low-affinity, high-capacity transporter, its presence on the BBM under high-load conditions allows rapid, high-volume absorption that SGLT1 alone (high affinity but lower capacity) cannot achieve
- This GLUT2 recruitment to the BBM also facilitates fructose absorption - since SGLT1 cannot transport fructose, the apical GLUT2 provides an extra route for fructose entry beyond GLUT5
- Evidence from SGLT1 knockout mice confirms this: Sglt1-null mice cannot recruit Glut2 to the BBM, confirming SGLT1-mediated entry as the trigger
Summary Table: The Three Intestinal Sugar Transporters
| Transporter | Gene | Membrane | Substrates | Mechanism | Na⁺ Dependent? | Affinity |
|---|---|---|---|---|---|---|
| SGLT1 | SLC5A1 | Apical (BBM) | Glucose, Galactose | Secondary active (2Na⁺:1 sugar) | Yes | High |
| GLUT5 | SLC2A5 | Apical (BBM) | Fructose only | Facilitated diffusion | No | - |
| GLUT2 | SLC2A2 | Basolateral (BLM); also BBM at high load | Glucose, Galactose, Fructose | Facilitated diffusion | No | Low |
Knockout Mouse Evidence
| Gene Deleted | Effect |
|---|---|
| Sglt1 | Glucose/galactose malabsorption; no fructose effect; no GLUT2 trafficking to BBM |
| Glut5 | Fructose malabsorption; glucose/galactose unaffected |
| Glut2 | Surprisingly, no defect in intestinal glucose absorption (suggesting alternative exit mechanisms like exocytosis); however lethal systemically due to loss of pancreatic β-cell glucose sensing → hypoinsulinemia and hyperglycemia |
Some Fructose Becomes Glucose Inside the Enterocyte
A portion of fructose entering the cell is:
- Phosphorylated
- Converted to glucose intracellularly
- Transported as glucose into the portal circulation
This is why fructose contributes to hepatic glucose/triglyceride load.
Clinical Relevance
| Condition | Mechanism |
|---|---|
| Glucose-Galactose Malabsorption | Rare autosomal recessive mutation in SGLT1 (SLC5A1); severe osmotic diarrhea in neonates on glucose/galactose feeds; responds to fructose-only diet |
| Fructose malabsorption | Reduced GLUT5 expression or capacity; bloating, diarrhea after fructose intake |
| SGLT2 inhibitors (gliflozins) | Act on SGLT2 in the kidney (not intestine) to block renal glucose reabsorption; diabetes treatment |
| Lactose intolerance | Deficient brush-border lactase; undigested lactose reaches colon → osmotic diarrhea, hydrogen gas (basis of breath hydrogen test) |
Sources: Guyton and Hall Textbook of Medical Physiology; Sleisenger and Fordtran's Gastrointestinal and Liver Disease; Sabiston Textbook of Surgery; Yamada's Textbook of Gastroenterology 7th ed.
This is a shared conversation. Sign in to Orris to start your own chat.