explain me about the anti gout drugs and reason behind the gout and macanism of action of the anti gout drugs
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Loading Image
Reading File
Loading Image
Reading File
I now have all the content needed for a complete, well-illustrated answer. Let me compile everything.
Gout: Pathogenesis and Anti-Gout Drugs
1. What is Gout?
Gout is a metabolic disorder characterized by abnormally high levels of uric acid in the blood (hyperuricemia, defined as serum uric acid > 6.8 mg/dL). This leads to deposition of monosodium urate (MSU) crystals in joints, soft tissues, and kidneys, triggering recurrent attacks of acute inflammatory arthritis.
- It is the most common form of inflammatory arthritis in adults.
- Primary gout accounts for ~90% of cases; secondary gout is caused by an underlying disease or medication.
2. Why Does Gout Happen? (Pathogenesis)
Step 1 - Uric Acid Overload
Uric acid is the end product of purine catabolism in humans. Purines (from DNA, RNA, and diet) are broken down via:
- De novo pathway - purine nucleotides synthesized from non-purine precursors
- Salvage pathway - free purines recycled by the enzyme HGPRT (hypoxanthine-guanine phosphoribosyltransferase)
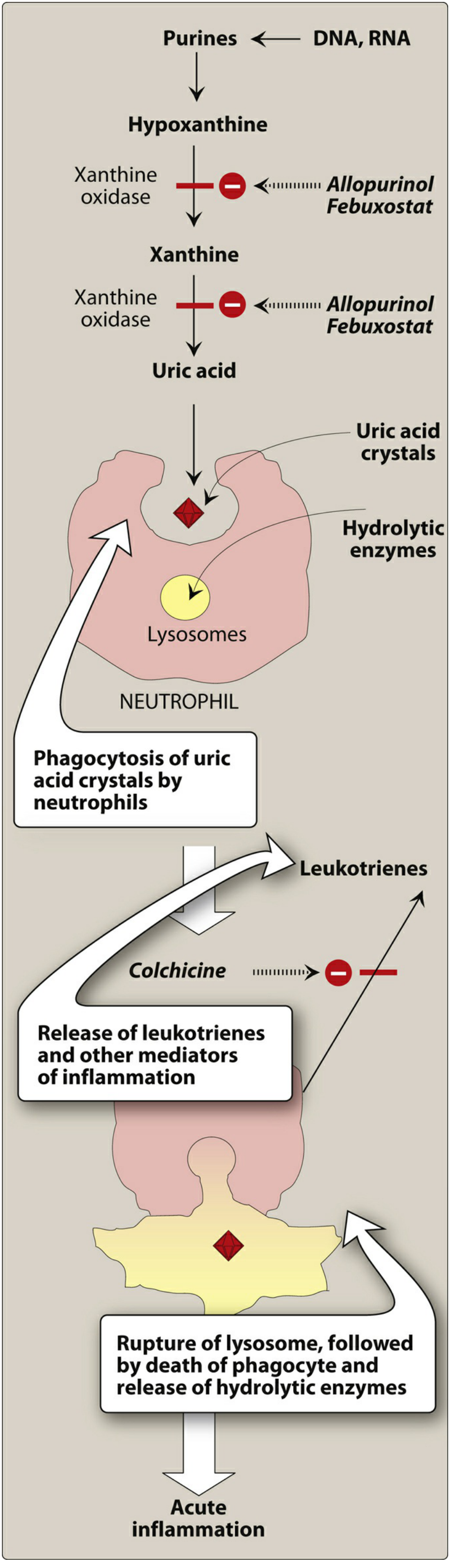
The enzyme xanthine oxidase catalyzes the final two steps:
Hypoxanthine → Xanthine → Uric Acid
Hyperuricemia arises from:
| Mechanism | Examples |
|---|---|
| Reduced excretion (most common, ~90%) | Idiopathic, chronic renal disease, diuretics (thiazides, furosemide), cyclosporine, aspirin |
| Overproduction | Enzyme defects (partial HGPRT deficiency), tumor lysis syndrome, leukemia, high-purine diet, alcohol |
| Both | Combined defects |
Key genetic links: Polymorphisms in URAT1, GLUT9, and KCNQ1 (urate transporters) have been identified as risk factors for primary gout.
Special syndromes:
- Lesch-Nyhan syndrome - complete HGPRT deficiency → severe hyperuricemia + neurological features (self-mutilation, intellectual disability)
Step 2 - Crystal Deposition and Inflammation
Once serum urate is high enough and the right conditions exist (lower temperature, lower pH in peripheral joints), MSU crystals precipitate. Here is the inflammatory cascade:
- Resident macrophages in the synovium phagocytose urate crystals
- Crystals activate the NLRP3 inflammasome (a cytosolic sensor)
- Inflammasome activates caspase-1, producing active IL-1β
- IL-1β recruits neutrophils into the joint
- Neutrophils phagocytose more crystals; crystals rupture their phagolysosomes, releasing lysosomal hydrolytic enzymes and free radicals
- Result: acute arthritis (classically the first metatarsophalangeal joint = podagra)
Repeated attacks lead to tophi (aggregates of urate crystals + inflammatory tissue) and permanent joint damage.
Diagram: Role of Uric Acid in Gout Inflammation
3. Clinical Phases of Gout
| Phase | Description |
|---|---|
| Asymptomatic hyperuricemia | Uric acid > 6.8 mg/dL, no symptoms; lasts 20-30 years before first attack |
| Acute gouty arthritis | Sudden, severe joint pain, redness, warmth; classically the big toe |
| Intercritical gout | Asymptomatic periods between attacks |
| Chronic tophaceous gout | Persistent deposits (tophi) in joints, ear pinnae, tendons; joint destruction |
4. Anti-Gout Drugs
Anti-gout drugs fall into two groups:
- Acute attack drugs - treat the inflammation rapidly
- Urate-lowering drugs - prevent future attacks by reducing uric acid
A. Drugs for ACUTE Gout
1. NSAIDs (e.g., Indomethacin)
Mechanism: Inhibit cyclooxygenase (COX-1 and COX-2) enzymes, reducing prostaglandin synthesis, which decreases pain and inflammation.
- Drug of choice for acute gout in patients without contraindications
- Indomethacin is the classic NSAID used
- All NSAIDs are generally effective
- Adverse effects: GI irritation, renal toxicity, increased CV risk, bleeding
2. Colchicine
Mechanism:
- Colchicine is a plant alkaloid that binds to tubulin (a microtubular protein) and causes its depolymerization
- This disrupts neutrophil motility, preventing their migration into the inflamed joint
- Also blocks cell division by binding to mitotic spindles
- It does NOT lower uric acid - it is purely anti-inflammatory
Clinical use:
- Must be given within 36 hours of attack onset to be effective
- Relieves pain within 12 hours
- Also used prophylactically during initiation of urate-lowering therapy (to prevent flares triggered by rapid urate changes)
Pharmacokinetics:
- Oral; absorbed rapidly from GI tract
- Metabolized by hepatic CYP3A4
- Undergoes enterohepatic recirculation
- Dose adjustment needed with CYP3A4 inhibitors (e.g., clarithromycin, itraconazole) and P-gp inhibitors (e.g., amiodarone, verapamil)
Adverse effects:
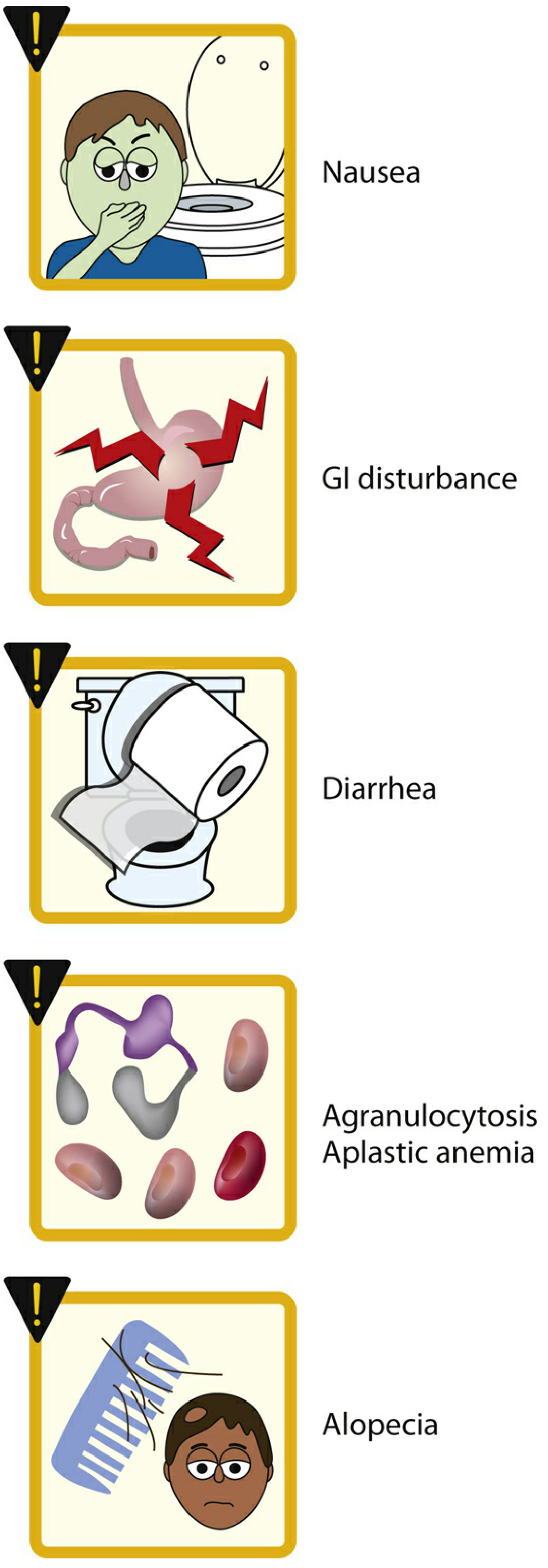
- Nausea, vomiting, abdominal pain, diarrhea (most common)
- Chronic use: myopathy, neutropenia, aplastic anemia, alopecia
- Contraindicated in pregnancy; use with caution in hepatic/renal/cardiovascular disease
3. Corticosteroids
Mechanism: Broad anti-inflammatory action via inhibition of phospholipase A2 (blocks prostaglandin and leukotriene synthesis at the source).
- Intra-articular injection for 1-2 joints affected
- Systemic corticosteroids (oral/IV) for polyarticular or severe attacks
- Useful when NSAIDs and colchicine are contraindicated
B. Drugs for CHRONIC Gout (Urate-Lowering Therapy)
Prophylactic urate-lowering therapy is indicated when:
- More than 2 gouty attacks per year
- Presence of tophi
- Chronic kidney disease, kidney stones
Important: Starting urate-lowering therapy can itself trigger an acute attack due to rapid shifts in serum urate. Always co-prescribe low-dose colchicine or NSAIDs for at least 6 months when initiating.
4. Allopurinol (First-line)
Mechanism:
- A purine analog that acts as a competitive inhibitor of xanthine oxidase
- Blocks the conversion of: Hypoxanthine → Xanthine → Uric Acid
- Its active metabolite, alloxanthine (oxypurinol), also inhibits xanthine oxidase with a half-life of 15-18 hours, allowing once-daily dosing
Uses: Gout, hyperuricemia from malignancy (tumor lysis syndrome), renal disease
Pharmacokinetics:
- Completely absorbed orally
- Metabolized to alloxanthine (also active)
- Excreted in urine; dose reduction required if GFR < 30 mL/min/1.73 m²
Adverse effects:
- Hypersensitivity (skin rash) - most common; risk increased with renal impairment
- Allopurinol hypersensitivity syndrome (rare but severe): fever, rash, hepatitis, renal failure
- Drug interaction: increases toxicity of 6-mercaptopurine and azathioprine (since xanthine oxidase also metabolizes these drugs)
5. Febuxostat
Mechanism: Non-purine xanthine oxidase inhibitor, structurally unrelated to allopurinol. Blocks the same enzyme (hypoxanthine → xanthine → uric acid).
Differences from allopurinol:
- Lower risk of rash/hypersensitivity reactions
- Less renal elimination - less dose adjustment needed in renal impairment
- Reserved for patients with contraindications to or intolerance of allopurinol
- Caution in patients with heart disease or stroke history - associated with higher cardiovascular risk vs. allopurinol
6. Probenecid (Uricosuric Agent)
Mechanism:
- A weak organic acid that inhibits the urate-anion exchanger (URAT1) in the proximal renal tubule
- At therapeutic doses, it blocks tubular reabsorption of uric acid, increasing its urinary excretion
- Does NOT reduce uric acid production
Adverse effects: Nausea, vomiting, dermatologic reactions, rarely anemia or anaphylaxis
Contraindication: Avoid if creatinine clearance < 50 mL/min (reduced excretion cannot compensate)
Note: Probenecid is an alternative for patients intolerant to or inadequately responding to xanthine oxidase inhibitors.
7. Pegloticase
Mechanism:
- A recombinant form of urate oxidase (uricase)
- Converts uric acid → allantoin (a water-soluble, non-toxic metabolite excreted by the kidneys)
- Humans lack endogenous uricase; most other mammals use this enzyme to metabolize uric acid
Use: Reserved for patients with refractory gout who fail standard therapies
Administration: IV infusion every 2 weeks
Adverse effects:
- Infusion-related reactions and anaphylaxis
- Premedication with antihistamines and corticosteroids required
5. Summary Table
| Drug | Class | Mechanism | Used For |
|---|---|---|---|
| NSAIDs (Indomethacin) | Anti-inflammatory | COX inhibition → ↓ prostaglandins | Acute attack |
| Colchicine | Anti-inflammatory | Tubulin depolymerization → ↓ neutrophil migration | Acute attack + prophylaxis |
| Corticosteroids | Anti-inflammatory | Phospholipase A2 inhibition | Acute attack (when NSAIDs/colchicine CI) |
| Allopurinol | Xanthine oxidase inhibitor | ↓ Uric acid synthesis (competitive) | Chronic gout (1st line) |
| Febuxostat | Xanthine oxidase inhibitor | ↓ Uric acid synthesis | Chronic gout (2nd line) |
| Probenecid | Uricosuric | ↑ Renal uric acid excretion (blocks URAT1) | Chronic gout (alternative) |
| Pegloticase | Recombinant uricase | Converts uric acid → allantoin | Refractory gout |
Sources: Lippincott Illustrated Reviews: Pharmacology, Robbins & Kumar Basic Pathology, Robbins, Cotran & Kumar Pathologic Basis of Disease
This is a shared conversation. Sign in to Orris to start your own chat.