seminar on generalised choroidal dystrophies and hereditary vitreoretinal degenerations including albinism
Reading File
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Loading Image
Loading Image
Loading Image
Loading Image
I now have comprehensive material for a full seminar. Let me compile everything.
Seminar: Generalised Choroidal Dystrophies, Hereditary Vitreoretinal Degenerations, and Albinism
Source: Kanski's Clinical Ophthalmology, 10th Edition - Chapter 15
Part 1: Introduction and Basic Science
General Framework
The inherited retinal degenerations ('retinal dystrophies') are a group of disorders exerting their major effect on the RPE-photoreceptor complex and choriocapillaris, causing a range of visual impairment. Hereditary retinal diseases are now the leading cause of blindness certification in the working-age population (age 16-64) in the UK.
Key genetic principles:
- Mutations in more than 250 genes have been linked to inherited retinal degenerations (full gene table: RetNet database)
- Genetic heterogeneity: mutations in different genes can cause the same clinical phenotype
- Phenotypic heterogeneity: the same gene can cause different phenotypes
- Genetic sequencing is mandatory in every patient with a suspected IRD
Photoreceptor Anatomy
| Feature | Rods | Cones |
|---|---|---|
| Number | 120 million | 6 million |
| Distribution | Dense outside macula | Concentrated at fovea |
| Function | Night/peripheral/motion vision | Day, colour, central vision |
| Types | One type | L (red), M (green), S (blue, ~2%) |
| Dysfunction symptoms | Nyctalopia, field loss | Poor central VA, dyschromatopsia |
Each photoreceptor has an outer segment (light-sensitive, contains visual pigment discs) and an inner segment (metabolic, contains mitochondria). Disc renewal - disc shedding from the outer segment tip and phagocytosis by RPE - is continuous and critical; failure underpins many dystrophies.
Part 2: Generalised Choroidal Dystrophies
2.1 Choroideremia
Inheritance: X-linked (gene: CHM)
- Males predominantly affected; clinically significant disease is almost exclusively in males
- Female carriers: mild patchy peripheral RPE mottling; VA, fields and ERG usually normal
- A contiguous extended gene deletion can also cause deafness and mental handicap
Pathology: Progressive, diffuse degeneration of the choroid, RPE and photoreceptors
Symptoms: Nyctalopia from adolescence → reduced peripheral vision → central vision loss (late)
Fundus signs - staged progression:
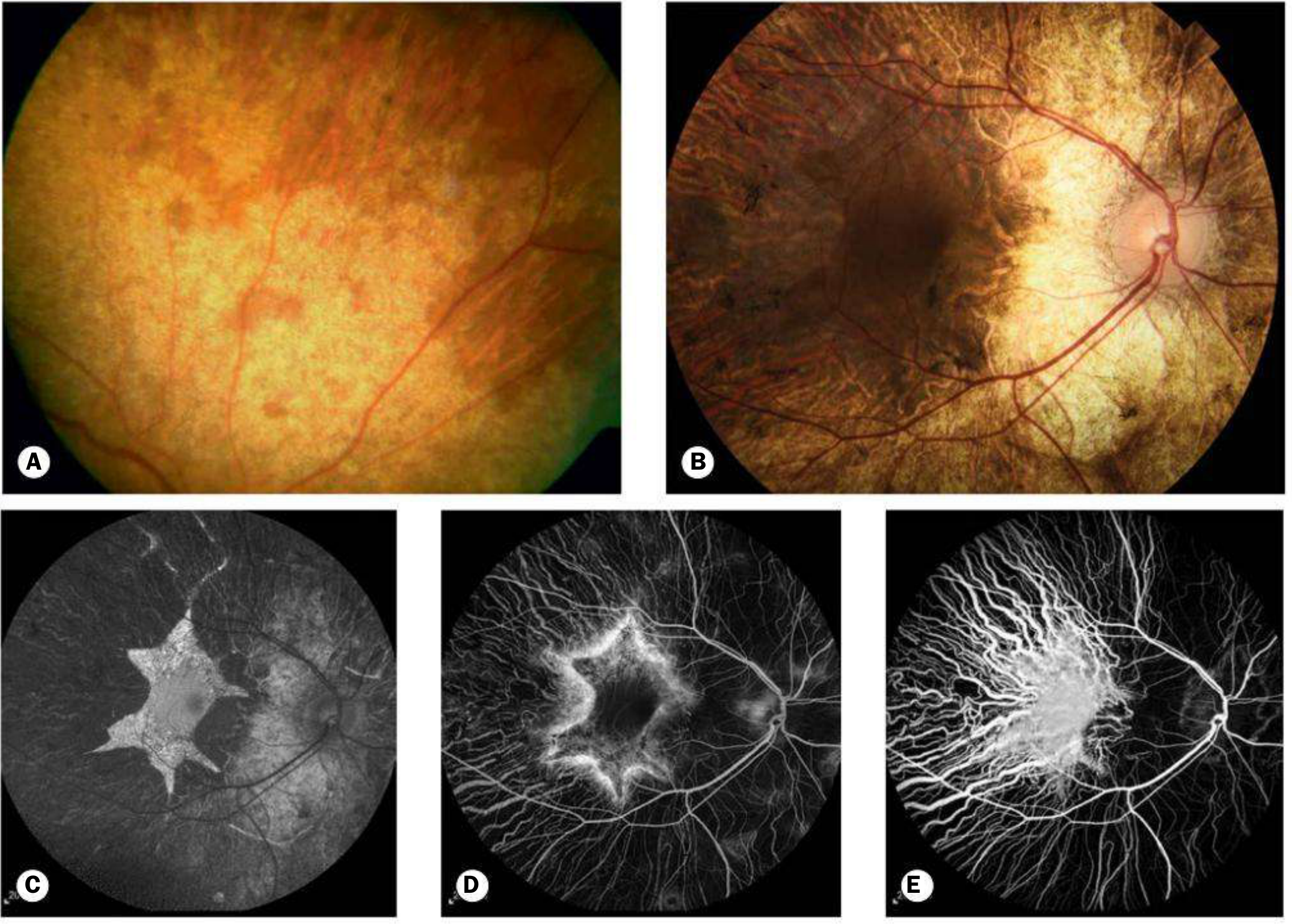
Fig. 15.35 Choroideremia: (A) Female carrier with subtle peripheral mottling; (B) advanced male disease with chorioretinal atrophy spreading centrally but fovea relatively spared until late; (C) FAF; (D) FA; (E) ICGA
- Carriers: patchy peripheral RPE atrophy and mottling (Fig. 15.35A)
- Males (early): mid-peripheral RPE abnormalities resembling RP
- Males (progress): chorioretinal atrophy spreading peripherally AND centrally
- End-stage: isolated choroidal vessels coursing over bare sclera
Key differentiator from primary retinal dystrophies: the fovea is relatively spared until late, allowing reasonable central vision well into middle age.
Investigations:
- FAF: characteristic scalloped border with hypoAF centrally and hyperAF at border
- ERG: progressively subnormal
Prognosis: Very poor. Most retain some vision until the sixth decade, then severe visual loss.
Genetics/Therapy: CHM gene therapy trials are ongoing. Gene identification is important for trial enrolment.
2.2 Gyrate Atrophy
Inheritance: AR (gene: OAT, chromosome 10q)
Pathophysiology: Deficiency of ornithine aminotransferase → elevated ornithine in plasma, urine, CSF and aqueous humour. This accumulation is toxic to the RPE and photoreceptors.
Symptoms: Myopia and nyctalopia in adolescence → gradual worsening
Fundus signs:
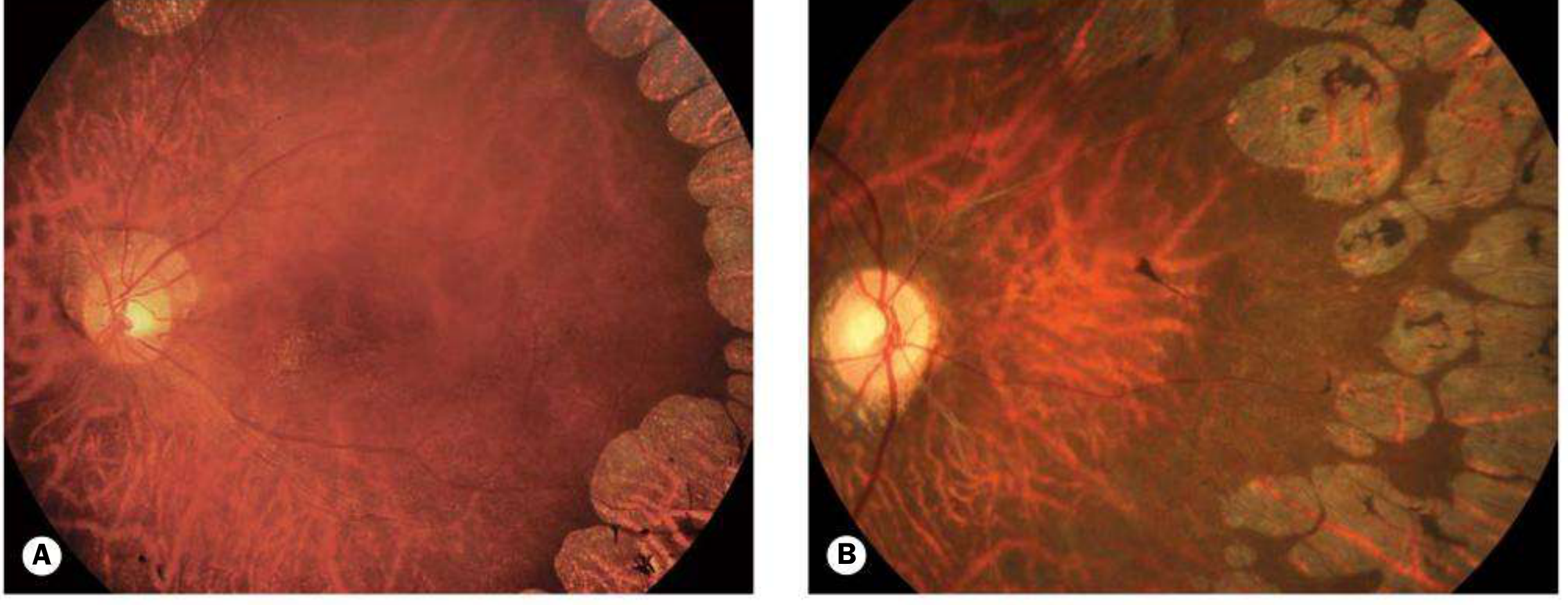
Fig. 15.36 Gyrate atrophy: (A) early disease - sharply demarcated circular/oval areas of chorioretinal atrophy; (B) advanced disease with peripheral and central coalescence
- Mid-peripheral depigmented spots + diffuse pigmentary mottling (asymptomatic phase)
- Sharply demarcated circular/oval chorioretinal atrophic areas - highly characteristic
- Glistening crystals at the posterior pole
- Coalescence with progressive peripheral and central spread; fovea spared until late
- Posterior subcapsular and posterior sutural cataracts by the second decade
- Vitreous degeneration, CMO and ERM may also occur
Investigation:
- Serum ornithine levels (confirms diagnosis)
- FA: sharp demarcation between areas of choroidal atrophy and normal choriocapillaris
- ERG: subnormal early → extinguished late
Treatment:
- No cure
- Arginine-restricted diet can slow progression (difficult adherence)
- Vitamin B6 (pyridoxine) supplementation reduces plasma ornithine in some patients
- Cataract surgery may provide transient visual improvement
Prognosis: Legal blindness around age 50
2.3 Central Areolar Choroidal Dystrophy (CACD)
Inheritance: Genetically heterogeneous, typically AD
Presentation: Third to fourth decades, gradual central visual impairment
Progression:
- Non-specific foveal granularity
- Well-circumscribed RPE atrophy + loss of choriocapillaris
- Slowly expanding geographic atrophy with prominence of large choroidal vessels
Investigation: OCT, FAF
Treatment: None available. Prognosis for central vision is poor.
2.4 Progressive Bifocal Chorioretinal Atrophy
A rare AD condition with progressive chorioretinal atrophy beginning in the first months of life as a temporal and nasal focus, which gradually extends to involve the entire fundus. Nystagmus and poor vision are early features.
Part 3: Retinitis Pigmentosa and Atypical Forms
3.1 Retinitis Pigmentosa (RP) - Classic
Definition: Clinically and genetically diverse group of inherited diffuse retinal degenerative diseases initially predominantly affecting rod photoreceptors, with subsequent degeneration of cones and RPE.
Prevalence: Most common hereditary retinal degeneration - 1:3000 to 1:5000
Inheritance patterns:
| Pattern | Severity | Notes |
|---|---|---|
| AD | Best prognosis | Most due to rhodopsin gene mutations |
| AR | Intermediate | Genetically most heterogeneous |
| X-linked | Most severe (6/60 by 5th decade) | 90% due to RPGR gene mutations |
| Sporadic | Variable | May be any of the above |
Genetic defect: Mutations in >100 gene loci for non-syndromic RP. About half of affected individuals have no identified molecular genetic abnormality. Gene defects affect: (a) phototransduction cascade, (b) retinoid cycle, (c) photoreceptor structure, (d) other biological functions of photoreceptor/RPE.
Classic diagnostic triad:
- Bone-spicule retinal pigmentation (perivascular, mid-peripheral)
- Arteriolar attenuation
- 'Waxy' disc pallor
Symptoms (in order of onset):
- Nyctalopia and dark adaptation difficulties (early, often presenting symptom)
- Peripheral visual field loss
- Photopsia (not uncommon)
- Reduced central vision (late, or earlier if cataract develops)
- Colour vision affected as cones involved
Additional fundus signs:
(This image is actually from juvenile retinoschisis - see section below)
- Bilateral mid-peripheral intraretinal perivascular bone-spicule pigmentary changes
- RPE atrophy
- Macula: atrophy, ERM, CMO (detected in ~15% on OCT)
- Optic disc drusen occur more commonly in RP patients
- Myopia is common
X-linked carrier females:
- Normal fundi OR golden-metallic ('tapetal') reflex at macula
- Centrifugal hyperautofluorescent lines on FAF (most striking feature)
- Small peripheral bone-spicule patches
RP sine pigmento: Absence/paucity of pigment - not a separate disease; every RP patient is initially sine pigmento as bone spicules develop secondary to degeneration.
Complications: Posterior subcapsular cataract (common), open-angle glaucoma (3%), PVD
Investigations:
- FAF: abnormal perimacular ring of hyperAF (increased lipofuscin from RPE dysfunction) + patchy hypoAF in mid-periphery. Distinguishes RP from normal in 95% of cases as a stand-alone test
- OCT: macular oedema in ~15%, macular atrophy in ~40%
- Visual fields: mid-peripheral scotomas → ring scotoma → tunnel vision → extinguished
- Full-field ERG: reduced scotopic rod response in early disease; eventually extinguished (sensitive but rarely needed clinically once diagnosis established)
- Dark adaptation test: prolonged (useful in equivocal early cases)
- Genetic analysis: identifies specific mutation; enables genetic counselling and trial enrolment
Management:
- Luxturna (voretigene neparvovec): approved gene therapy for RPE65 mutations
- No other specific commercial treatment available; gene therapy trials ongoing
- Annual follow-up for treatable complications (cataract, CMO)
- CMO: carbonic anhydrase inhibitors (topical or systemic)
- Cataract surgery is safe and appropriate
3.2 Atypical RP - Syndromic Forms (20-30% of cases)
Usher Syndrome (AR, genetically heterogeneous)
- Accounts for ~5% of all profound deafness in children; ~50% of all combined deafness and blindness
- Type I (MYO7A-associated, 75%): profound congenital sensorineural deafness + severe RP with extinguished ERG in first decade; vestibular dysfunction
- Type II (most common in ophthalmic practice): moderate hearing loss, later onset RP
- Type III (2%): progressive hearing loss + late-onset RP
- Cochlear implants can treat Type I deafness; gene therapy trials (MYO7A) underway
Bassen-Kornzweig Syndrome (Abetalipoproteinemia, AR)
- Dysfunctional fat and fat-soluble vitamin (A, D, E, K) absorption
- Failure to thrive → severe spinocerebellar ataxia
- Blood film: acanthocytosis ('thorny' red cells)
- Fundus: scattered white dots → RP-like changes by end of first decade
- Other: ptosis, ophthalmoplegia, strabismus, nystagmus
- Treatment: vitamin supplementation + low-fat diet
Refsum Disease (AR) - Adult/Infantile Forms
- Phytanic acid accumulation throughout body
- Skin changes (ichthyosis), neurological and visceral features
- Retinal changes: RP-like or 'salt and pepper'
- Cataract, optic atrophy may occur
- Treatment: low phytanic acid diet retards progression
Bardet-Biedl Syndrome (AR, genetically heterogeneous)
- Mutations in genes for proteins involved in non-motor cilia function (ciliopathy)
- Systemic features: learning disability, polydactyly, obesity, renal abnormalities, hypogonadism
- Retinal: rod-cone dystrophy with bull's eye maculopathy pattern
- Management: multidisciplinary; gene-specific therapy under investigation
Neuronal Ceroid Lipofuscinoses (NCL)
- Group of lysosomal storage diseases with accumulation of ceroid lipofuscin
- Features: progressive neurodegeneration, dementia, seizures, retinal degeneration
3.3 Enhanced S-cone Syndrome and Goldmann-Favre Syndrome
Gene: NR2E3 (encodes ligand-dependent transcription factor) - AR, variable expressivity
Pathophysiology: Hyperfuction of S-cones + severe impairment of M- and L-cones; non-recordable rod function. Goldmann-Favre is the severe variant of the same gene mutation.
Symptoms: Nyctalopia in childhood ± hemeralopia; decreased colour vision (tritan axis preserved)
Signs:
- Pigmentary changes along vascular arcades or mid-periphery, with round pigment clumps
- Cystoid maculopathy (without fluorescein leakage)
- Vitreous degeneration and peripheral retinoschisis
OCT: Disrupted foveal lamination with thickened subfoveal ellipsoid zone
ERG: No rod response; scotopic maximum = photopic (both show S-cone waveform)
Prognosis: Variable; ~30% have VA 6/36 or worse by late middle age
Part 4: Hereditary Vitreoretinal Degenerations
4.1 Juvenile X-linked Retinoschisis
Inheritance: X-linked (gene: RS1, Xp22.13 - encodes retinoschisin)
Pathophysiology: Retinoschisin is secreted by photoreceptors; involved in intercellular adhesion. The basic defect involves splitting of the retinal nerve fibre layer from the rest of the sensory retina (contrast with acquired retinoschisis which splits at outer plexiform layer).
Epidemiology: Rare congenital disease; presents in boys age 5-10 years with difficulty reading; less commonly squint/nystagmus in infancy.
Carrier females: Asymptomatic, no retinal changes
Signs:
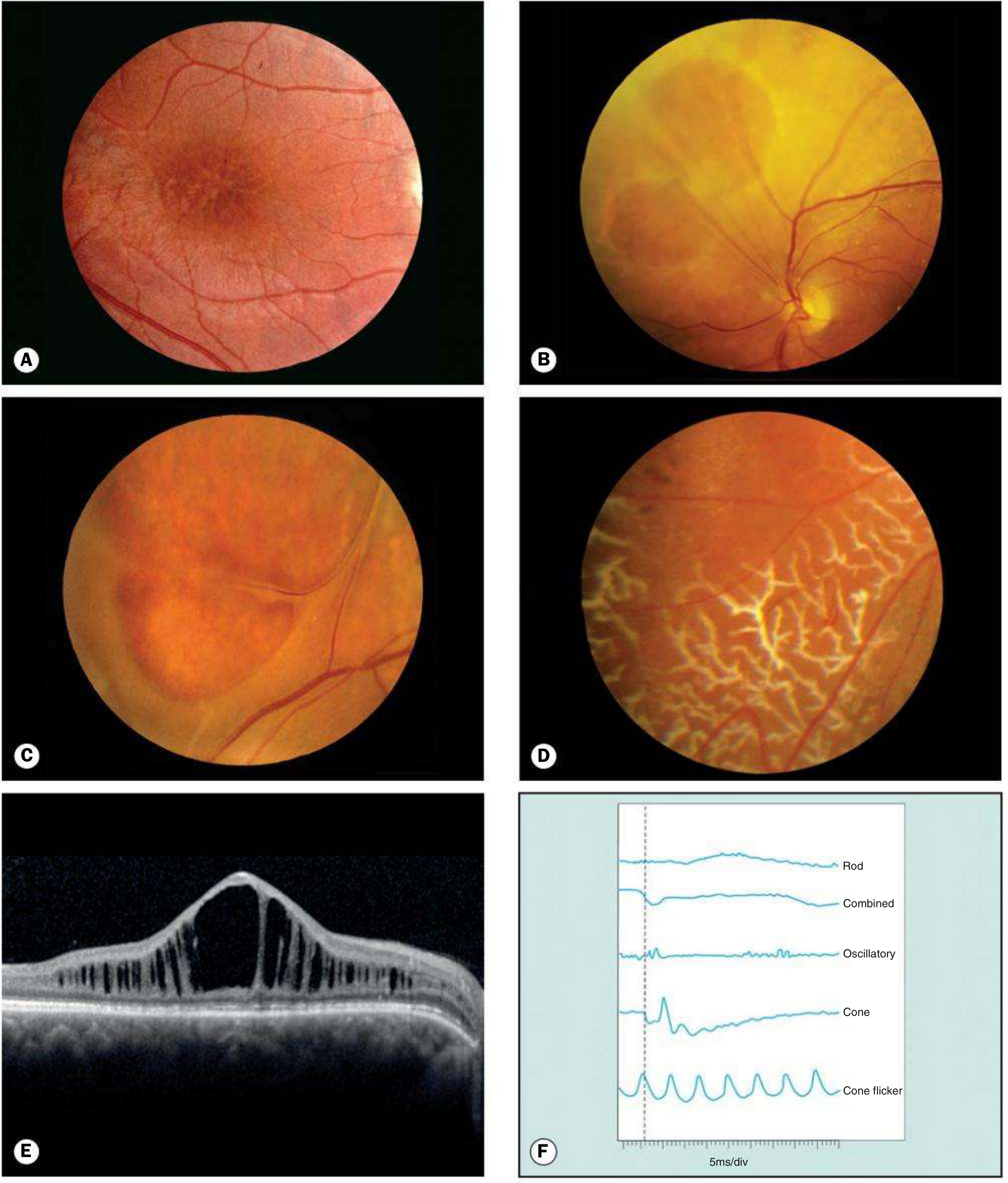
Fig. 15.38 Juvenile retinoschisis: (A) 'Bicycle wheel' spoke-like foveal schisis; (B) large oval inner leaf defects; (C) 'vitreous veils'; (D) peripheral silvery dendritic figures; (E) OCT with cysts in inner nuclear/outer plexiform layers; (F) ERG with negative a-wave and reduced b-wave
- Foveal schisis (most common): spoke-like striae radiating from foveola with cystoid changes - the "bicycle wheel" appearance
- White drusen-like dots and pigment variation
- Peripheral schisis: infero-temporal quadrant predominantly; inner layer (ILM + nerve fibre layer) develops oval defects → coalesce → 'vitreous veils' (retinal blood vessels floating in vitreous)
- Silvery peripheral dendritic figures, vascular sheathing, retinal flecks
Complications: Vitreous/intra-schisis haemorrhage, neovascularisation, subretinal exudation, rarely RD (rhegmatogenous or tractional)
Investigations:
- FAF: spoke-like patterns, central hypoAF with surrounding hyperAF
- OCT: cystic spaces in inner nuclear and outer plexiform layers
- ERG: characteristic negative ERG - reduced b-wave amplitude with preserved a-wave (reflects inner retinal dysfunction); scotopic more affected than photopic
- Genetic testing: RS1 mutations
Prognosis: VA deteriorates in first two decades then may stabilise until fifth/sixth decade. Vitreous haemorrhage and RD are the main causes of significant visual loss.
Treatment:
- Carbonic anhydrase inhibitors (topical dorzolamide or oral acetazolamide) may reduce foveal schisis
- Vitreoretinal surgery for RD or non-clearing vitreous haemorrhage
- RS1 gene therapy trials are underway
4.2 Stickler Syndrome (Hereditary Arthro-ophthalmopathy)
Genetics: Genetically heterogeneous collagen connective tissue disorder; 8 sub-groups; mutation identified in 95% of cases
- STL1 (AD, most common): COL2A1 mutations; membranous vitreous type
- STL2 (AD): COL11A1 mutations; beaded vitreous type
- STL3 (AD): COL11A2; no ocular features (non-ocular type)
- STL4-8 (AR): extremely rare
Epidemiology: ~1 in 7500-9000. Most common inherited cause of retinal detachment in children
Systemic features:
- Mid-facial hypoplasia
- Pierre-Robin sequence: micrognathia, cleft palate, glossoptosis
- Bifid uvula
- Joint hypermobility, spondyloepiphyseal dysplasia, early-onset osteoarthritis
- Sensorineural or conductive deafness (recurrent otitis media)
TIP: Consider Stickler syndrome in any infant with cleft palate + deafness + joint hypermobility + myopia
Ocular features (three characteristic):
- High myopia (congenital, non-progressive)
- Vitreoretinal degeneration with extremely high rate of retinal detachment
- Cataract
Vitreous signs:
- STL1: optically empty vitreous + retrolenticular membrane + circumferential equatorial vitreous membranes
- STL2: fibrillary and beaded vitreous appearance
Retinal signs:
- Radial lattice-like degeneration with RPE hyperplasia
- Risk of giant retinal tear and RD - leading cause of childhood blindness in this syndrome
Management:
- Prophylactic laser/cryotherapy to lattice degeneration and tears
- Prompt vitreoretinal surgery for RD
- Multidisciplinary: hearing aids, orthodontics, orthopaedics, genetics
- Annual retinal surveillance from diagnosis
4.3 Wagner Syndrome (VCAN-related Vitreoretinopathy)
Inheritance: AD (gene: VCAN - versican)
Note: Erosive vitreoretinopathy is now known to be the same disorder
Key distinction from Stickler: No systemic abnormalities
Signs:
- Low to moderate myopia
- Optically empty vitreous cavity (lacking structural elements - reduced 'scaffolding')
- Greyish-white avascular strands/membranes extending into vitreous
- Circumferential ridge-like condensation at or anterior to retinal periphery
- Progressive peripheral chorioretinal atrophy
- Deficient peripheral retinal vasculature
- Nyctalopia (commonly troublesome); progressive field constriction
- Cataract (common in younger adults); glaucoma
Investigations:
- FA: non-perfusion due to choriocapillaris loss
- ERG: initially normal → reduction of scotopic b-wave → diffuse abnormality
Complications: Retinal detachment in up to 50%, often before age 15
4.4 Snowflake Vitreoretinal Degeneration
Inheritance: AD (gene: KCNJ13) - rare; some similarities to Wagner syndrome
Stages (I-IV) - characteristic snowflake deposits:
- Stage I: Early; fibrillar vitreous condensation
- Stage II: Snowflake deposits developing (minute crystalline yellow-white dots)
- Stage III: Retinal vessel sheathing
- Stage IV: Advanced with RPE changes
Prognosis: Retinal detachment less common than Wagner; prognosis usually very good
Part 5: Albinism
5.1 Introduction
Albinism is a genetically determined heterogeneous group of disorders of melanin synthesis affecting either:
- Eyes alone = Ocular albinism (OA)
- Eyes + skin + hair = Oculocutaneous albinism (OCA)
OCA is subdivided into:
- Tyrosinase-negative (complete; no melanin at all)
- Tyrosinase-positive (incomplete; variable melanin)
Molecular genetics: 7 subtypes of non-syndromic OCA (OCA 1-7)
Key unifying mechanism: Different mutations act through a common pathway involving reduced melanin synthesis in the eye during development, leading to:
- Foveal hypoplasia
- Iris transillumination
- Nystagmus
- Misrouting of optic nerve fibres at the chiasm (excess crossing)
Important: Patients with oculocutaneous albinism have increased risk of cutaneous basal cell and squamous cell carcinoma - sun protection is mandatory.
5.2 Tyrosinase-negative OCA (OCA Type 1)
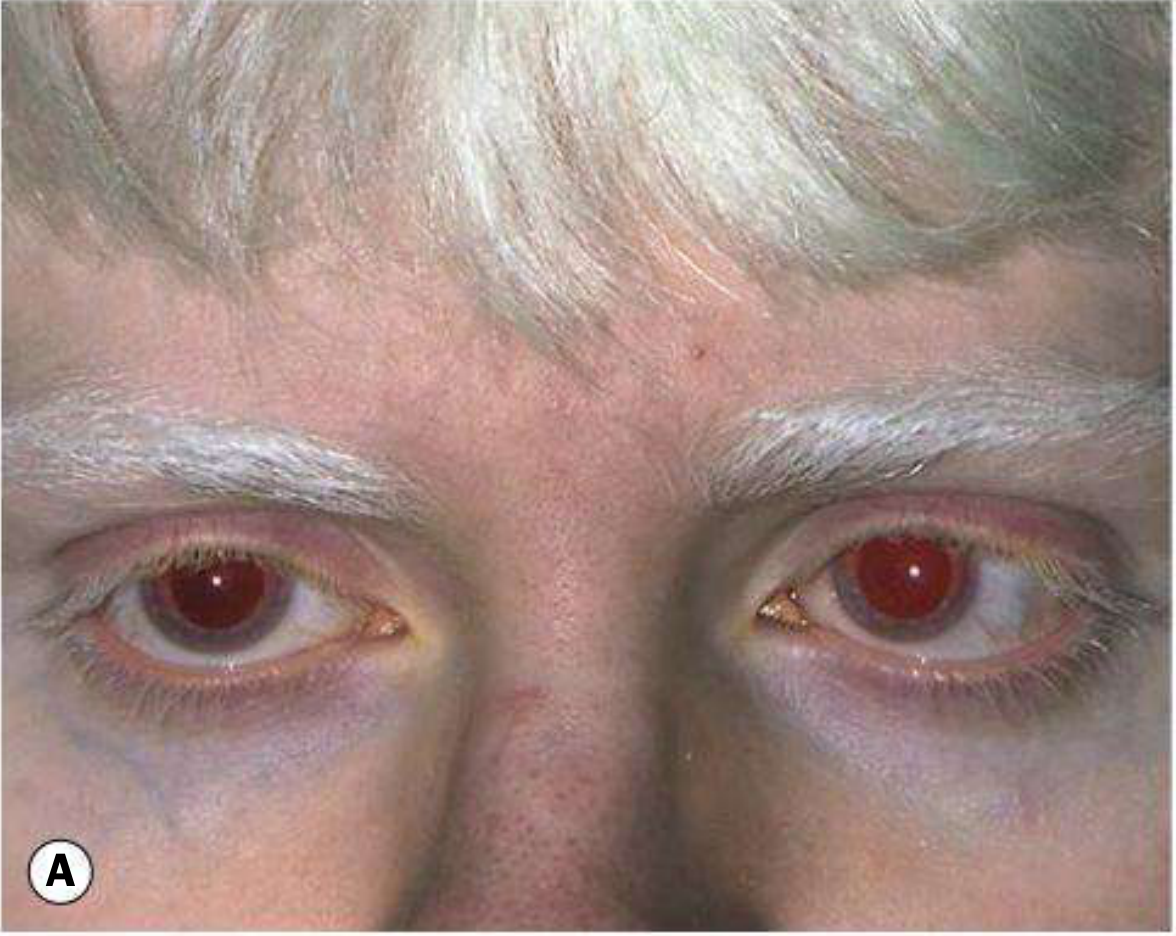
Fig. 15.44A - OCA type 1: white hair, pale skin in tyrosinase-negative OCA patient
Gene: TYR (chromosome 11q14) - encodes tyrosinase; AR inheritance
Phenotype: Incapable of synthesising ANY melanin - white hair, very pale skin throughout life
Ocular signs:
- VA usually <6/60 due to foveal hypoplasia
- Pendular horizontal nystagmus - typically increases in bright illumination; lessens with age
- Diaphanous, translucent iris - 'pink-eyed' appearance (entire iris diaphanous on retroillumination)
- Fundus: totally lacks pigment; conspicuously large choroidal vessels; foveal hypoplasia with absence of foveal pit and poorly formed perimacular vascular arcades
- Chiasmal misrouting: majority of fibres from each eye cross to the contralateral hemisphere (more than normal) - demonstrable with visual evoked potentials (asymmetric VEPs are the electrophysiological signature)
- High refractive errors, positive angle kappa, squint, absent stereopsis
5.3 Tyrosinase-positive OCA (OCA Type 2)
Gene: OCA2 (chromosome 15q12) - plays role in tyrosine transport in melanosomes; AR; hundreds of mutations
Epidemiology: Most common form in Africa; accounts for one-third of cases worldwide
Phenotype: Synthesise variable amounts of melanin; hair may be white/yellow/red, darkens with age; skin pale at birth but usually darkens by age 2
Ocular signs:
- VA usually impaired due to foveal hypoplasia
- Iris may be blue or dark brown with variable translucency
- Fundus: variable hypopigmentation
- Nystagmus and refractive errors common
5.4 OCA Types 3-7
- OCA type 3 (TYRP1 gene): seen especially in Black Africans
- Types 4-7: extremely rare, reported in single families
5.5 Systemic Associations of Albinism
Chediak-Higashi syndrome: Rare AR disorder - mutation of a lysosomal regulator protein → failure of phagolysosome formation → recurrent pyogenic infections (especially Staphylococcus); partial albinism, neurological features, lymphoma-like accelerated phase
Hermansky-Pudlak syndrome: AR lysosomal storage disease with platelet dysfunction (easy bruising, excessive bleeding); in some cases pulmonary fibrosis and granulomatous colitis with bleeding - potentially life-threatening
Waardenburg syndrome: AD condition with:
- White forelock, poliosis
- Synophrys ('monobrow')
- Sensorineural deafness
- Sometimes limb abnormalities
- Iris heterochromia
5.6 Ocular Albinism (OA)
Inheritance: X-linked (OA1; GPR143 gene)
Key distinction: Skin and hair pigmentation is normal (or near-normal); ocular features predominate
Carrier females: Normal vision but show characteristic iris translucency and a mottled/mud-splattered fundal appearance (due to X-inactivation mosaic)
Males: Full ocular phenotype - nystagmus, foveal hypoplasia, reduced VA, iris transillumination, fundus hypopigmentation, chiasmal misrouting
Part 6: Investigations Summary
Electrophysiology
| Condition | ERG pattern |
|---|---|
| RP (early) | Reduced scotopic rod response, combined response |
| RP (advanced) | Extinguished all responses |
| XLRS (juvenile retinoschisis) | Negative ERG (reduced b-wave, preserved a-wave) |
| Enhanced S-cone syndrome | No rod response; scotopic max = photopic (both S-cone shaped) |
| Gyrate atrophy (early) | Subnormal |
| Choroideremia (advanced) | Subnormal to extinguished |
| Albinism | Asymmetric VEPs (chiasmal misrouting) |
Multimodal Imaging
| Modality | Key use |
|---|---|
| FAF | RP (perimacular ring), choroideremia (scalloped borders), XLRS |
| OCT | Macular oedema/atrophy in RP; cystic spaces in XLRS; foveal schisis |
| FA | Choroidal atrophy in gyrate, Wagner, choroideremia |
| Full-field ERG | Sensitive for early/equivocal RP; diagnostic for XLRS |
| Dark adaptation test | Early RP, equivocal cases |
| VEP | Albinism (misrouting) |
Part 7: Management Principles
Gene Therapy - Current Status
| Condition | Gene | Therapy Status |
|---|---|---|
| RP with RPE65 mutations | RPE65 | Luxturna - FDA/EMA approved, commercially available |
| Choroideremia | CHM | Phase II/III trials ongoing |
| XLRS | RS1 | Phase I/II trials |
| Usher Syndrome Type I | MYO7A | Trials ongoing |
Medical/Supportive Management
- RP: Vitamin A palmitate (historically used, evidence debated); annual review; low vision aids; genetic counselling
- Gyrate atrophy: Arginine-restricted diet; Vitamin B6; cataract surgery
- RP macular oedema: Topical/oral carbonic anhydrase inhibitors (acetazolamide)
- XLRS macular schisis: Topical CAIs (dorzolamide) to reduce cystic spaces
- Albinism: Tinted lenses/photochromic lenses for photophobia; refraction correction; low vision rehabilitation; mandatory sun protection (photocarcinogenesis risk); strabismus surgery
Genetic Counselling
All inherited retinal degenerations require:
- Pedigree construction
- Molecular genetic testing to identify causative mutation
- Counselling regarding transmission risks
- Identification of potential trial eligibility
- Cascade testing of family members
Quick Reference: Key Differentials
| Feature | Choroideremia | Gyrate Atrophy | RP |
|---|---|---|---|
| Inheritance | X-linked | AR | AD/AR/X-linked |
| Gene | CHM | OAT | >100 genes |
| Biochemical marker | None | Elevated ornithine | None |
| Atrophy pattern | From periphery, scalloped | Discrete oval areas coalesce | Mid-peripheral pigment |
| Foveal sparing | Until late | Until late | May be involved earlier |
| ERG | Subnormal → extinguished | Subnormal → extinguished | Rod reduced → extinguished |
| Treatment | Gene therapy (trial) | Low-arg diet, Vit B6 | Luxturna (RPE65 only) |
Source: Kanski's Clinical Ophthalmology: A Systematic Approach, 10th Edition, Chapter 15 - Inherited Retinal Degenerations. RetNet gene database lists all currently known IRD genes.
This is a shared conversation. Sign in to Orris to start your own chat.