Aml pathology simple language
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
I now have all the content I need for a clear, simple-language explanation of AML pathology. Here is the full answer:
Acute Myeloid Leukemia (AML) - Pathology in Simple Language
(Source: Robbins, Cotran & Kumar - Pathologic Basis of Disease)
What Is AML?
Think of your bone marrow as a factory that makes blood cells. Normally, immature blood cells (called "blasts") grow up and mature into useful cells - red cells, white cells, platelets. In AML, some of these immature myeloid cells get stuck in their baby stage. They have mutations that say "stop maturing" and "keep multiplying." These stuck, immature cells are called myeloblasts, and they flood the bone marrow and blood.
The word breakdown: Acute = happens fast | Myeloid = from the myeloid cell line | Leukemia = cancer of blood cells.
Who Gets It?
- Can happen at any age, but gets more common as you get older
- Peaks after age 60
- About 13,000 new cases per year in the United States
What Goes Wrong? (Pathogenesis)
Mutations in the leukemia cells fall into four basic types of "damage":
| Category | What It Means in Simple Terms |
|---|---|
| Transcription factor mutations | The "instruction manuals" for cell maturation get scrambled. Cells can't read how to grow up. |
| Receptor/signaling mutations | Cells get constant "grow! grow! grow!" signals that never shut off (e.g. FLT3 mutations) |
| Epigenetic mutations | The "dimmer switch" on genes gets broken (IDH1/2, DNMT3A, TET2 mutations) |
| Tumor suppressor mutations | The "brakes" on cell division get cut (e.g. TP53 mutations) |
Usually, at least one mutation from each of the first two categories is needed to cause AML. No single mutation alone is enough.
Microscope Findings (Morphology)
Diagnosis requires ≥ 20% blasts in the bone marrow (except in some genetic subtypes where even fewer blasts confirm the diagnosis).
What the blasts look like under the microscope:
- Large cells with delicate, open chromatin (compared to lymphoblasts)
- Prominent nucleoli (2-4 per cell)
- More cytoplasm than lymphoblasts
- Azurophilic (pink-purple) granules in the cytoplasm
- Auer rods - these are the hallmark! They look like pink needles inside the cell. They are clumped granules fused together. They are found only in myeloid leukemia, not lymphoid.
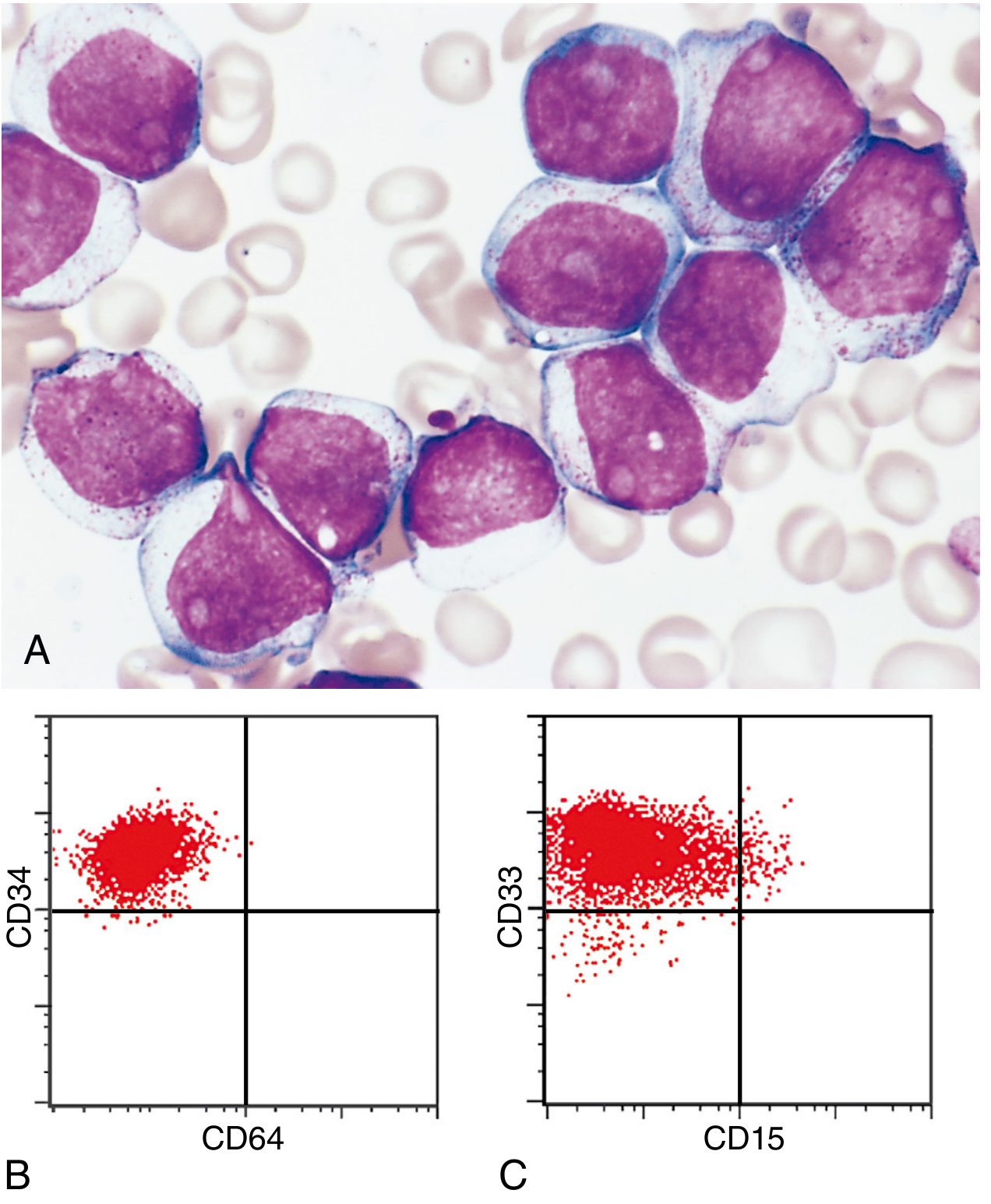
Fig 13.31 (A) Myeloblasts with open chromatin, prominent nucleoli, and fine azurophilic granules. (B-C) Flow cytometry: blasts are CD34+ (stem cell marker) and CD33+ (immature myeloid marker), but CD64-negative (not yet mature). - Robbins Pathology
Different Blast Types
| Blast Type | Key Feature |
|---|---|
| Myeloblasts | Granules in cytoplasm, Auer rods present |
| Monoblasts | Folded/lobulated nucleus, no Auer rods, positive for nonspecific esterase |
| Megakaryoblasts | Show platelet-lineage features, often with marrow fibrosis |
| Erythroblasts | Rare; linked to TP53 mutations |
Note: Sometimes zero blasts are found in the blood ("aleukemic leukemia"). This is why a bone marrow biopsy is essential in any patient with unexplained low blood counts.
Subtypes and Genetics
AML is classified by its genetic/chromosomal changes. These determine prognosis and treatment:
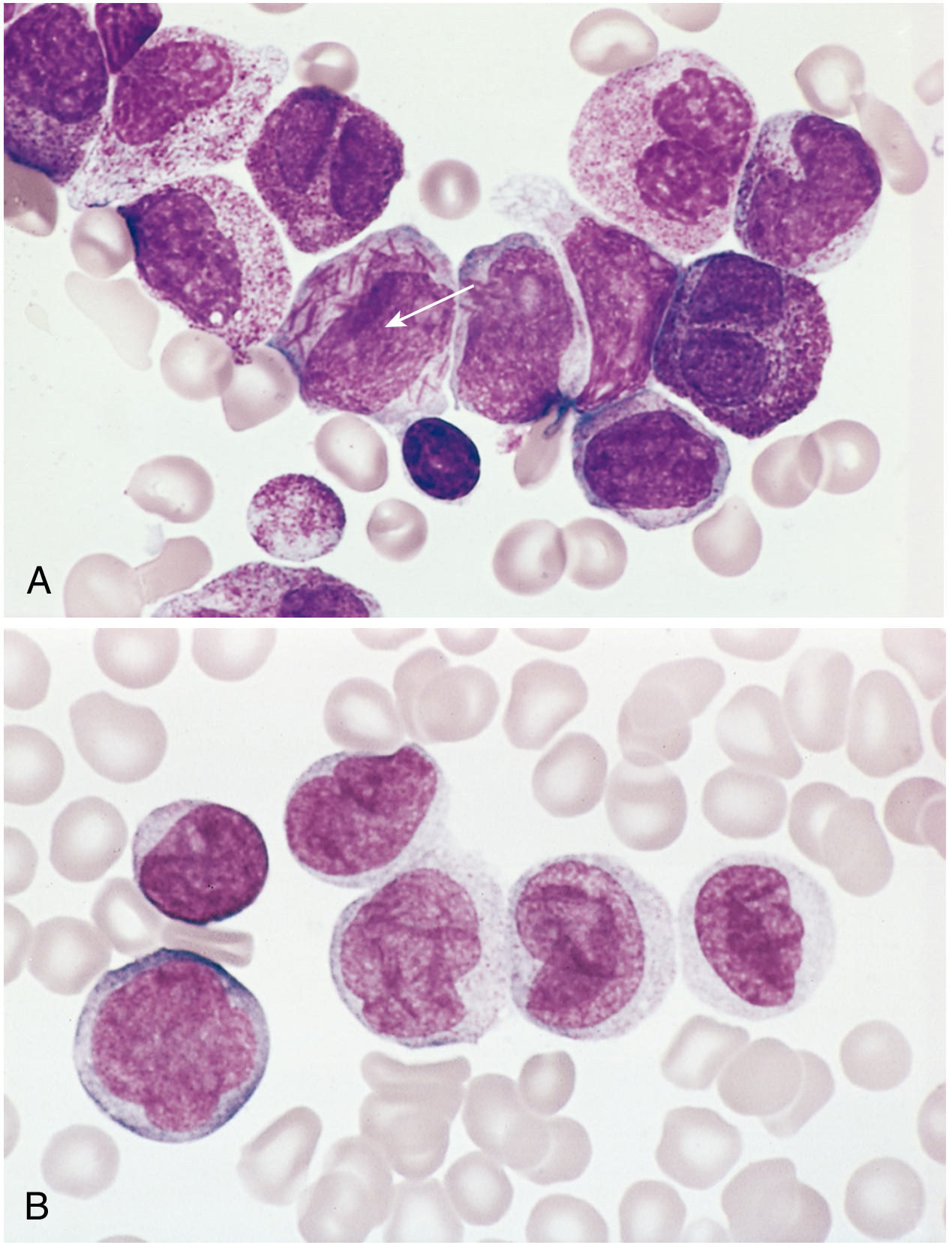
Fig 13.32 AML subtypes: (A) Acute promyelocytic leukemia - note the cell packed with Auer rods (arrow). (B) Monocytic AML - cells with folded kidney-shaped nuclei. - Robbins Pathology
Major genetic subtypes:
| Genetics | Prognosis | Key Point |
|---|---|---|
| t(8;21) - RUNX1::RUNX1T1 | Good | Auer rods easy to find |
| inv(16) - CBFB::MYH11 | Good | Mix of myeloid + monocytic cells, abnormal eosinophils |
| t(15;17) - PML::RARA | Very Good (>90% cure) | Acute Promyelocytic Leukemia (APL); tons of Auer rods in bundles; danger: can cause DIC; responds to ATRA + arsenic trioxide |
| KMT2A rearrangement t(11q23) | Poor | Monocytic features, common in infants |
| NPM1 mutation | Good | Common; detected by sequencing |
| FLT3 mutation | Poor (without targeted therapy) | Treated with FLT3 inhibitors |
| IDH1/IDH2 mutation | Variable | IDH inhibitors now available |
| TP53 mutation | Very Poor | Often from chemo/radiation exposure |
Two major categories in the WHO classification:
- AML with specific genetic aberrations (genetics define the diagnosis)
- AML defined by differentiation (no specific genetic marker found)
Immunophenotype (What Markers the Cells Show)
Flow cytometry is used to confirm AML and distinguish it from ALL (lymphoid leukemia):
- CD34 - positive (stem cell marker; cells are still very immature)
- CD33 - positive (immature myeloid marker)
- CD64 - negative (absent because cells haven't matured enough)
- CD15 - variably positive (present on more mature myeloid cells)
- MPO (myeloperoxidase) - positive (the key myeloid marker; negative in lymphoblasts)
How AML Destroys the Body (Consequences)
The blasts flood the marrow and crowd out normal blood cell production, causing:
| Problem | Cause | Symptoms |
|---|---|---|
| Anemia | No red cells being made | Fatigue, pallor, breathlessness |
| Thrombocytopenia | No platelets being made | Bruising, petechiae (tiny red dots on skin), gum bleeding, heavy periods |
| Neutropenia | No mature white cells | Infections (fungal, bacterial, opportunistic - Pseudomonas, Candida) |
Extra features:
- DIC (Disseminated Intravascular Coagulation) - especially in APL (t15;17); the leukemic cells release clotting factors that trigger body-wide clotting, then paradoxically cause catastrophic bleeding. This is a hematologic emergency.
- Leukemia cutis - monocytic AML cells infiltrate the skin, causing raised nodules
- Gingival hypertrophy - gum swelling, seen in monocytic subtypes (cells infiltrate gum tissue)
- Myeloid sarcoma - a solid lump of leukemia cells forming outside the bone marrow (rare)
- CNS involvement - less common than in ALL
Why AML Happens (Risk Factors)
- Radiation or chemotherapy exposure - causes deletions of chromosomes 5 and 7, or TP53 mutations
- Myelodysplastic syndrome (MDS) - "pre-leukemia" that can transform into AML
- Alkylating agent chemotherapy (e.g. cyclophosphamide, melphalan) - causes chromosome 5/7 deletions
- Topoisomerase II inhibitors (e.g. etoposide) - causes KMT2A rearrangements at 11q23
- Down syndrome - especially megakaryoblastic AML in children
Prognosis and Treatment (Simple Summary)
- Overall, about 60% achieve complete remission with chemotherapy
- Only 15-30% are disease-free at 5 years (overall)
- APL (t15;17) is the exception - curable in >90% with ATRA + arsenic trioxide (no conventional chemo needed)
- t(8;21) and inv(16) - relatively good with standard chemotherapy
- High-risk AML (elderly, post-MDS, post-chemo, TP53-mutated) - treated with bone marrow transplant (HSC transplantation) when possible
- New targeted therapies: FLT3 inhibitors (midostaurin, gilteritinib), IDH inhibitors (enasidenib, ivosidenib), venetoclax (BCL-2 inhibitor)
Summary Table
| Feature | AML in Simple Terms |
|---|---|
| What it is | Immature myeloid blood cells get stuck and pile up |
| Where | Bone marrow (flooded by blasts) |
| Diagnosis marker | Auer rods (pink needles inside cells) |
| Key blast count | ≥20% in marrow (or any count + specific genetics) |
| Confirms myeloid origin | MPO+, CD33+, CD34+, CD64- |
| Emergency subtype | APL (t15;17) - DIC risk |
| Best prognosis | APL (>90% cure with ATRA + arsenic) |
| Worst prognosis | TP53 mutations, post-MDS, elderly |
| Treatment backbone | Cytarabine + anthracycline ("7+3"), then targeted agents |
- Robbins, Cotran & Kumar - Pathologic Basis of Disease

17840230800508358143266518155476.jpg
Explain
analyse_media
Finding Sources
Finding Sources
Reading File
I have everything needed. Here is a clear, simple explanation of the entire note:
CML Pathology - Full Explanation of Your Notes
Your note covers Chronic Myeloid Leukemia (CML) - its molecular genetics, how the disease starts, what goes wrong in the cell, and how it differs from AML.
Part 1 - Myelopoiesis (Normal Blood Cell Development)
The top of the note shows the normal maturation sequence of myeloid cells:
Myeloblast → Promyelocyte → Myelocyte → Metamyelocyte → Band forms → Segmented forms → Mature Neutrophils
This is the normal factory line. In CML, this line still works (cells can still mature) - but the factory is running at 10x speed.
Part 2 - Molecular Genetics (The Root Cause)
This is the most important part. CML is caused by one specific chromosomal swap:
The Two Chromosomes Involved:
| Chromosome | Gene It Carries | What That Gene Does |
|---|---|---|
| Chromosome 9 (long arm) | ABL gene | Makes a tyrosine kinase (an enzyme that adds phosphate to proteins). It also has a myristoyl binding site (MBS) - this acts as the "OFF switch" for the kinase |
| Chromosome 22 (long arm) | BCR gene | Breakpoint Cluster Region - acts as a "promoter" that drives gene activity |
The Translocation (the swap):
- A piece of chromosome 9 (containing ABL) breaks off and sticks onto chromosome 22 (near the BCR gene)
- Specifically: the A2 exon of ABL fuses onto exon 13/14 of BCR
- This creates a fusion gene called BCR-ABL on chromosome 22
- Chromosome 22 now looks shorter than normal - this shortened chromosome 22 is called the Philadelphia (Ph) chromosome
- Found in 95% of CML cases
- This is a balanced reciprocal translocation - meaning chromosome 9 also gets a piece of chromosome 22 in exchange (though the chromosome 9 side is not clinically significant)
Simple analogy: Imagine gene ABL is a car with working brakes (the MBS). When it fuses to BCR, the brakes are removed. The car (kinase) now runs at full speed with no way to stop.
Part 3 - The Disease Mechanism (Step by Step)
BCR-ABL fusion gene is formed
↓
Myristoyl binding site (the "OFF switch") is LOST
↓
ABL kinase becomes CONSTITUTIVELY ACTIVE (always switched ON)
↓
Acts as a docking site for ATP (the fuel)
↓
Tyrosine phosphorylation happens non-stop
↓
Sends constant "GROW! DIVIDE! SURVIVE!" signals
↓
Dysregulated production of CLONAL hematopoietic stem cells
↓
Uncontrolled proliferation of granulocytes (→ Granulocytosis, Basophilia)
+ Platelets (→ Thrombocytosis)
+ RBCs
Key point: In CML, normal differentiation is preserved - blasts still mature into adult cells. So the blood is full of ALL stages of maturing white cells, not just blasts.
This massive overproduction causes the marrow to overflow into the spleen → Massive Splenomegaly (the hallmark of CML).
Part 4 - CML vs AML (The Key Comparison Table)
| Feature | CML | AML |
|---|---|---|
| Differentiation of myeloblasts | Fairly normal (cells mature) | Complete arrest (cells stuck as blasts) |
| Proliferation | High, multiple lineages (granulocytes + platelets + RBCs all increased) | Very high, but single lineage (only one type of blast) |
| Blood picture | Full spectrum of maturing cells | Predominantly immature blasts |
| Driver | BCR-ABL (tyrosine kinase) | Various mutations blocking differentiation |
Why does this matter? In CML, because cells mature, the patient can survive for years in the "chronic phase." In AML, blast arrest causes rapid marrow failure and the patient gets sick very fast.
Part 5 - Treatment: Tyrosine Kinase Inhibitors (TKIs)
Since the entire disease is driven by one overactive enzyme (BCR-ABL), you can target it directly with drugs:
| Drug | How It Works |
|---|---|
| Imatinib (Gleevec) | Blocks the ATP-binding pocket of BCR-ABL - without ATP, the kinase can't phosphorylate anything, so the "grow" signal stops |
| Asciminib | A newer drug that works differently - it binds to the myristoyl binding site (MBS) - essentially restoring the "OFF switch" that was lost in the translocation |
This is why CML was one of the first cancers to be "tamed" by targeted therapy - one gene, one drug.
Summary in One Paragraph
CML starts when chromosomes 9 and 22 swap pieces, fusing the ABL gene onto BCR. This creates the BCR-ABL fusion protein that has lost its regulatory OFF-switch (the myristoyl binding site). The kinase is now permanently ON, sending non-stop growth signals to a hematopoietic stem cell. Unlike AML (where cells can't mature), in CML cells still mature - but there are too many of them, flooding the blood with granulocytes, platelets, and causing massive splenomegaly. The disease is treated with TKIs like imatinib that directly block the BCR-ABL kinase.
(Goldman-Cecil Medicine; Robbins Pathologic Basis of Disease)
This is a shared conversation. Sign in to Orris to start your own chat.