Factors affecting bioavailability of drug
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Reading File
I now have comprehensive information from both Katzung's and Lippincott's Pharmacology. Let me compile the full answer.
Factors Affecting Bioavailability of a Drug
Bioavailability is defined as the fraction of unchanged drug reaching the systemic circulation following administration by any route. For an intravenous (IV) dose, bioavailability = 100% by definition. For other routes (especially oral), it is commonly less than 100%.
Mathematically: F = f × (1 - ER)
where f = fraction absorbed from the gut, and ER = hepatic extraction ratio.
It is measured using the area under the plasma concentration-time curve (AUC):
Bioavailability = (AUC oral / AUC IV) × 100%
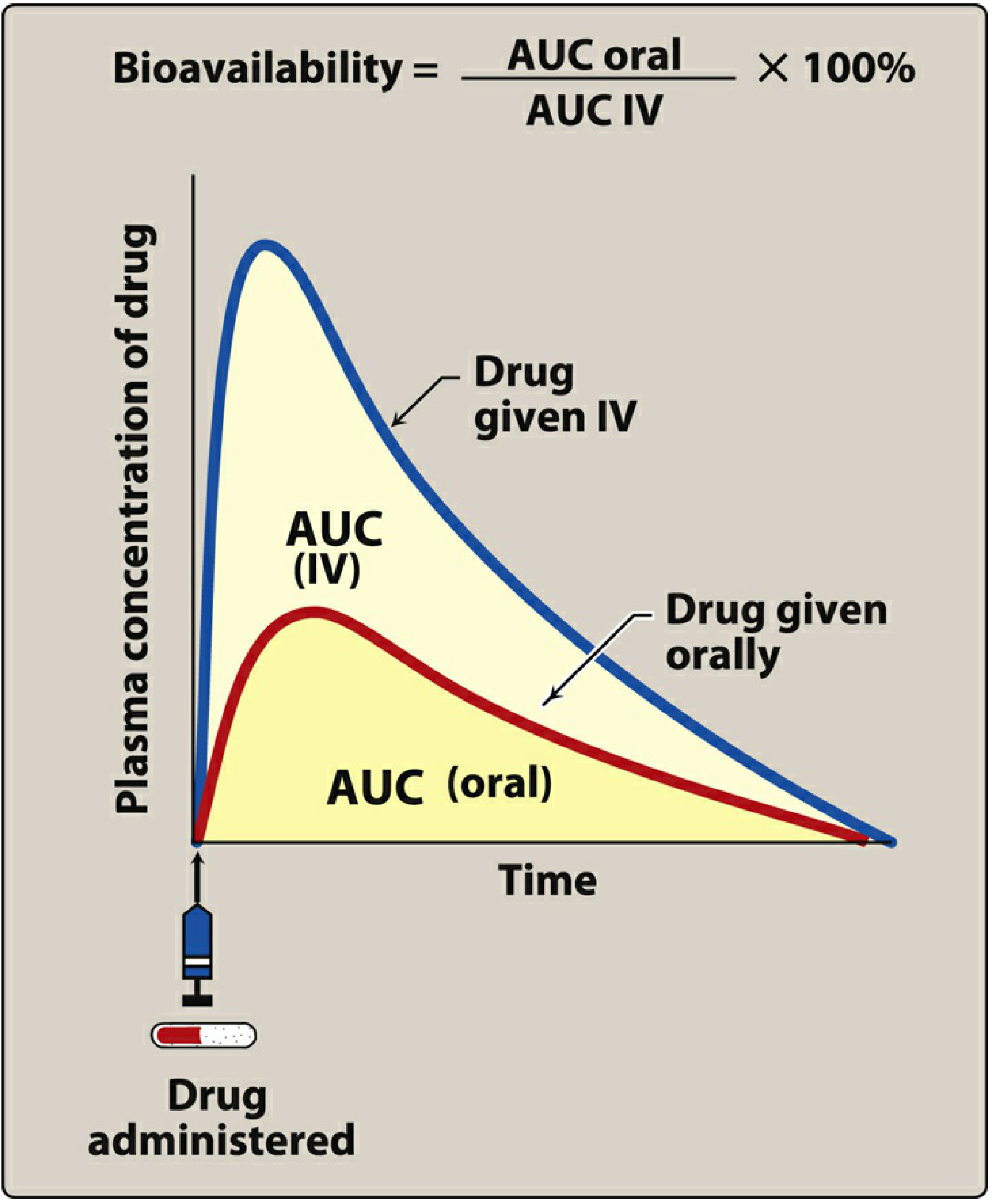
Factors Affecting Bioavailability
1. First-Pass Hepatic Metabolism (Most Important)
When a drug is absorbed from the GI tract, it travels via the portal circulation to the liver before entering systemic circulation. If the liver rapidly metabolizes the drug during this initial pass, the amount of active drug reaching the body is significantly reduced. This is called the first-pass effect (or first-pass elimination).
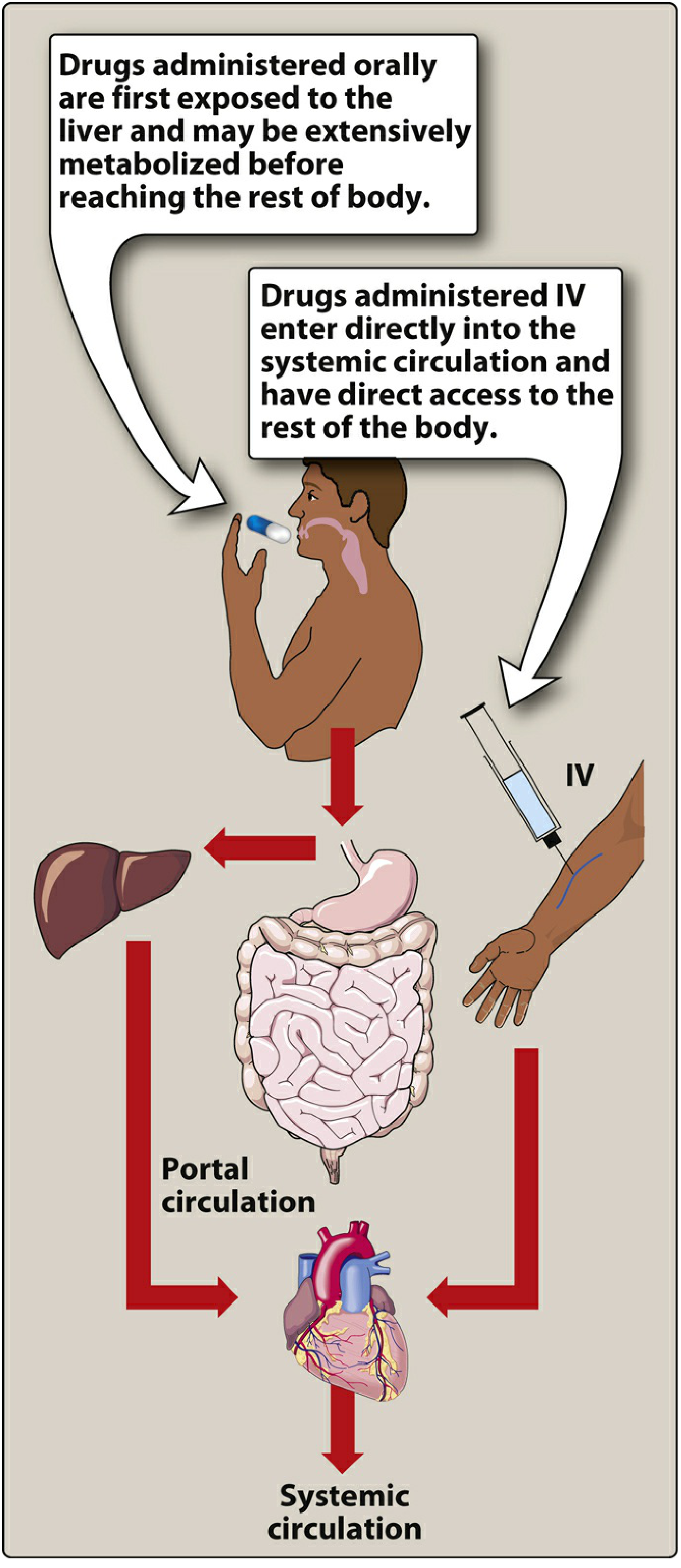
- Drugs with high extraction ratios have low oral bioavailability: morphine (~33%), propranolol, lidocaine, isoniazid, tricyclic antidepressants, nitroglycerin (>90% cleared)
- First-pass can also occur in the gut wall (e.g., via CYP3A4 enzymes) and in portal blood
- Hepatic blood flow affects this: in liver cirrhosis with portosystemic shunting, bioavailability increases for high-extraction drugs
The extraction ratio (ER) = hepatic clearance / hepatic blood flow (normal ~90 L/h in a 70 kg person).
Source: Katzung's Basic and Clinical Pharmacology, 16th Ed., p. 82; Lippincott Illustrated Reviews Pharmacology, p. 42
2. Solubility of the Drug
- Too hydrophilic (e.g., atenolol): cannot cross lipid-rich cell membranes - poorly absorbed
- Too lipophilic (e.g., acyclovir): insoluble in aqueous body fluids - cannot gain access to cell surfaces
- Ideal drugs are largely lipophilic with some aqueous solubility - this is why many drugs are weak acids or weak bases
- Very lipophilic drugs dissolve in the lipid bilayer but cannot move through it to the aqueous interior
Source: Katzung's 16th Ed., p. 82; Lippincott, p. 41
3. Route of Administration
The route determines what fraction reaches systemic circulation. Approximate bioavailabilities:
| Route | Bioavailability | Key Notes |
|---|---|---|
| Intravenous (IV) | 100% | By definition; most rapid |
| Intramuscular (IM) | 75 to ~100% | Large volumes feasible |
| Subcutaneous (SC) | 75 to ~100% | Smaller volumes |
| Transdermal | 80 to ~100% | Bypasses first-pass; prolonged absorption |
| Rectal (PR) | 30 to <100% | ~50% bypasses first-pass |
| Inhalation | 5 to <100% | Bypasses first-pass; rapid onset |
| Oral (PO) | 5 to <100% | First-pass effect significant |
| Sublingual | Near 100% | Directly enters systemic (not portal) veins |
Source: Katzung's 16th Ed., Table 3-3, p. 81
4. Drug Formulation (Pharmaceutical Factors)
- Particle size: smaller particles dissolve faster and are better absorbed
- Salt form: different salts of the same drug have different solubilities (e.g., erythromycin base vs. erythromycin stearate)
- Crystal vs. amorphous form: crystal forms may be more stable but less soluble
- Tablet coating and excipients: enteric coatings delay dissolution; poorly made tablets may not disintegrate properly
- These factors are why two products containing the same drug can have different bioavailabilities (bioequivalence issues)
Source: Lippincott, p. 41
5. Chemical Instability in the GI Tract
- Some drugs are destroyed by gastric acid pH: e.g., penicillin G is unstable in acidic stomach contents
- Others are degraded by GI tract enzymes: e.g., insulin is broken down by proteolytic enzymes
- Gut flora can also metabolize certain drugs before they are absorbed
Source: Lippincott, p. 41
6. P-Glycoprotein (Efflux Transporter)
- P-glycoprotein is a reverse transporter in the gut wall cells that actively pumps drug back into the gut lumen, reducing absorption
- This limits bioavailability of many drugs
- Inhibition of P-glycoprotein (e.g., by grapefruit juice) can substantially increase drug absorption and bioavailability
Source: Katzung's 16th Ed., p. 82
7. Gut Wall Metabolism (Pre-systemic Metabolism)
- Enzymes in the intestinal wall (notably CYP3A4) can metabolize drugs before they even reach the portal vein
- This is a component of first-pass elimination but distinct from hepatic first-pass
- Grapefruit juice inhibits intestinal CYP3A4, raising bioavailability of affected drugs
Source: Katzung's 16th Ed., p. 82
8. Splanchnic/Hepatic Blood Flow
- Hepatic blood flow determines the extraction ratio: higher flow can paradoxically decrease bioavailability by presenting more drug to metabolizing enzymes
- Liver disease (cirrhosis, portosystemic shunting): increases bioavailability of high-extraction drugs (e.g., propranolol)
- Congestive cardiac failure: reduced hepatic blood flow can unpredictably alter first-pass
Source: Katzung's 16th Ed., p. 83
9. GI Motility and Gastric Emptying Rate
- Rapid gastric emptying delivers drug faster to the small intestine (main absorption site) - can increase rate of absorption
- Slow motility (e.g., from opioids, anticholinergics): delays absorption
- Food can increase or decrease absorption depending on the drug; high-fat meals can increase absorption of lipophilic drugs
10. Drug Interactions
- Chelation and binding: antacids (containing Ca²+, Mg²+, Al³+) bind tetracyclines and fluoroquinolones, reducing absorption
- Enzyme inducers: rifampicin, phenytoin - increase intestinal and hepatic metabolism, reducing bioavailability
- Enzyme inhibitors: ketoconazole, erythromycin, grapefruit juice - reduce first-pass metabolism, increasing bioavailability
Summary Table
| Factor | Direction of Effect | Example |
|---|---|---|
| High first-pass hepatic metabolism | Decreases F | Morphine, propranolol, nitroglycerin |
| Extreme lipophilicity or hydrophilicity | Decreases F | Acyclovir (too lipophilic), atenolol (too hydrophilic) |
| P-glycoprotein efflux | Decreases F | Many CNS drugs |
| Sublingual/transdermal route | Increases F (bypasses first-pass) | Nitroglycerin sublingual |
| Acid instability | Decreases F | Penicillin G |
| Enzyme inhibitors | Increases F | Grapefruit juice + statins |
| Liver cirrhosis | Increases F (high-extraction drugs) | Propranolol in cirrhosis |
| Poor formulation (large particles) | Decreases F | Generic tablet vs. brand |
Key clinical takeaway: For drugs with high extraction ratios (e.g., propranolol, morphine, lidocaine), oral bioavailability is low AND highly variable between individuals because it depends on hepatic blood flow and inter-individual differences in CYP enzyme activity. This is why lidocaine is never given orally, and why nitroglycerin is given sublingually or transdermally rather than orally.
Primary sources: Katzung's Basic and Clinical Pharmacology, 16th Edition, pp. 81-83; Lippincott Illustrated Reviews Pharmacology, pp. 40-43
Is gastric emptying is an
It looks like your message got cut off! Could you complete your question?
Were you asking something like:
- "Is gastric emptying an important factor affecting bioavailability?"
- "Is gastric emptying an example of a pharmacokinetic factor?"
- Something else?
How does gastric emptying rate affect drug absorption?
Finding Sources
Reading File
Reading File
Reading File
Excellent - I have comprehensive, multi-textbook coverage. Here is the full answer:
How Gastric Emptying Rate Affects Drug Absorption
The Core Principle
The stomach is NOT a major site of drug absorption. The small intestine - with its enormous surface area and rich villous structure - is where most orally administered drugs are absorbed. Therefore, gastric emptying rate primarily determines how quickly a drug is delivered to the small intestine, which in turn determines the rate of absorption.
"Any factor that accelerates gastric emptying will generally increase the rate of drug absorption, whereas any factor that delays gastric emptying will have the opposite effect."
- Goodman & Gilman's Pharmacological Basis of Therapeutics, p. 46
Gastric Emptying and Rate vs. Extent of Absorption
| Parameter | Effect of Gastric Emptying |
|---|---|
| Rate of absorption (how fast) | Directly proportional - faster emptying = faster onset |
| Extent of absorption (how much total) | Usually NOT affected - total amount absorbed stays the same |
| Peak plasma concentration (Cmax) | Higher and earlier with faster emptying |
| Time to peak (Tmax) | Shorter with faster emptying |
This is clinically important: gastric emptying affects when a drug works, not usually how much of it works.
Special Case - Kinetics
When absorption rate is determined by gastric emptying (rather than passive diffusion), the mechanism is zero-order - i.e., the rate is independent of the drug amount remaining in the gut. Once the drug dissolves in GI fluids, absorption typically becomes first-order (rate proportional to concentration). - Katzung's Basic and Clinical Pharmacology, 16th Ed., p. 82
Factors That Influence Gastric Emptying Rate
According to Goodman & Gilman's, gastric emptying is influenced by:
- Food: Caloric content, volume, osmolality, temperature, and pH of ingested fluid all slow emptying
- Body position: Lying on the right side accelerates emptying
- Exercise vs. rest: Exercise generally slows emptying
- Diurnal variation: Emptying is faster in the morning
- Sex hormones: Estrogen slows gastric emptying - premenopausal women and those on estrogen replacement therapy have slower emptying than men
- Drugs (see below)
How Altered Gastric Emptying Changes Drug Behavior
Accelerated Gastric Emptying (faster delivery to intestine)
| Cause | Example |
|---|---|
| Prokinetic drug | Metoclopramide |
| Body position | Right lateral decubitus |
| Fasting state | - |
Effect: Faster onset of action; higher Cmax reached sooner. Particularly important for acid-unstable drugs - the faster the drug passes through the acidic stomach, the less it degrades, so absorption may actually be increased (both rate and extent). - Goldman-Cecil Medicine, p. 1656
Delayed Gastric Emptying (drug stays longer in stomach)
| Cause | Example |
|---|---|
| Food (especially high fat) | Large meal before dosing |
| Anticholinergic drugs | Atropine, tricyclic antidepressants |
| Opioids | Morphine, codeine |
| Lying on the left side | - |
| Diabetes (gastroparesis) | - |
| Hypothyroidism | - |
Effects:
- Delayed absorption onset - drug action is delayed
- Drug degradation by gastric acid - acid-labile drugs (e.g., penicillin G, some PPIs) may be destroyed before reaching the intestine
- Paradoxical benefit for poorly absorbed drugs - drugs like metformin that need more contact time with the absorbing surface may actually be better absorbed when intestinal motility is reduced by anticholinergics, giving the drug more time at the absorptive surface. - Goldman-Cecil Medicine, p. 1656
Clinical Examples
| Drug | Impact of Gastric Emptying |
|---|---|
| Paracetamol (acetaminophen) | Classic model - Tmax closely tracks gastric emptying rate; opioids delay its onset of analgesia |
| Levodopa | Delayed emptying allows more degradation in the stomach; metoclopramide improves its absorption |
| Penicillin G | Faster emptying = less acid degradation = more drug reaches intestine intact |
| Metformin | Reduced intestinal motility (anticholinergics) increases contact time and may increase absorption |
| Enteric-coated drugs (e.g., aspirin EC) | Must pass through stomach first; delayed emptying postpones dissolution and absorption |
Enteric Coatings and Controlled-Release Formulations
- Enteric-coated tablets are specifically designed to resist dissolution in gastric acid and only dissolve once they reach the alkaline environment of the small intestine. Delayed gastric emptying therefore delays the onset of action of enteric-coated drugs significantly.
- Controlled-release formulations deliberately use the principle of controlled delivery rate to smooth out absorption, essentially making the dosage form - not gastric emptying - the rate-limiting step. - Goodman & Gilman, p. 47
Summary
Gastric emptying rate is a rate-limiting factor for oral drug absorption. Faster emptying generally means faster absorption and earlier peak drug levels. It rarely affects the total amount absorbed - except in specific cases where acid lability or contact time with absorptive surfaces matters. Clinically, this explains why taking opioids alongside acetaminophen delays pain relief, why metoclopramide improves levodopa therapy, and why food-drug timing instructions matter for many medications.
Sources: Goodman & Gilman's Pharmacological Basis of Therapeutics, p. 46-47; Katzung's Basic and Clinical Pharmacology, 16th Ed., p. 82; Goldman-Cecil Medicine, p. 1656
This is a shared conversation. Sign in to Orris to start your own chat.