Atherosclerosis
Reading File
Finding Sources
Searching PubMed
"atherosclerosis"[MeSH Terms] AND "pathogenesis"[MeSH Terms]
Searching the Web
atherosclerosis pathophysiology mechanisms 2025 overview
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Reading File
Loading Image
I now have very rich content from Robbins and Guyton, plus two excellent diagrams. Let me compile the comprehensive answer.
Atherosclerosis
Definition
Atherosclerosis is best understood as the vascular response to endothelial injury. It is characterized by intimal lesions called atheromas (atherosclerotic plaques) - raised lesions with soft, lipid-rich cores (mainly cholesterol and cholesterol esters with necrotic debris) covered by fibrous caps. It affects large and intermediate-sized arteries and is the pathological basis for coronary, cerebral, and peripheral vascular disease, accounting for roughly half of all deaths in Western countries.
- Robbins & Kumar Basic Pathology, p. 313-319
Arteriosclerosis vs Atherosclerosis: Arteriosclerosis is a broader term referring to thickened and stiffened vessels of all sizes. Atherosclerosis is specifically lipid-driven, intima-based disease of large and medium arteries.
Epidemiology
-
Virtually ubiquitous in high-income nations; rapidly increasing in lower-income countries due to urbanization and Western diet adoption
-
Myocardial infarction is responsible for approximately one-quarter of all deaths in the United States
-
Death rates from coronary artery disease in Africa, India, and Southeast Asia now exceed those in the United States; eastern European countries have rates 3-5x higher than the US, and 7-12x higher than Japan
-
Risk factors are roughly multiplicative: two factors increase MI risk ~4-fold; hyperlipidemia + hypertension + smoking together increase the rate 7-fold (Framingham Heart Study data)
-
Robbins & Kumar Basic Pathology, p. 314
Risk Factors
| Category | Factors |
|---|---|
| Non-modifiable (Constitutional) | Family history, inherited causes (e.g., familial hypercholesterolemia), increasing age, male sex |
| Modifiable | Hyperlipidemia, hypertension, cigarette smoking, diabetes mellitus, inflammation |
Key constitutional factors:
- Genetics: Family history is the most important independent risk factor. Most familial risk is polygenic (related to HTN, DM), though single-gene disorders like familial hypercholesterolemia exist
- Age: Incidence of MI increases 5-fold between 40 and 60 years. Aging also drives clonal hematopoiesis of indeterminate potential (CHIP), which alters monocyte/macrophage function and contributes to atherogenesis
- Sex: Premenopausal women are relatively protected; after menopause the incidence can even exceed that in men
Key modifiable factors:
- Hyperlipidemia: The most important modifiable risk factor. Elevated LDL and reduced HDL are most relevant. Oxidized LDL is directly toxic to endothelium and drives macrophage foam cell formation. Statins (HMG-CoA reductase inhibitors) reduce LDL and significantly lower cardiovascular risk
- Hypertension: Increases risk 2-fold for IHD; mechanical shear stress injures the endothelium
- Cigarette smoking: Accelerates atherosclerosis via oxidative stress, endothelial injury, and pro-thrombotic effects
- Diabetes mellitus: Causes accelerated, diffuse atherosclerosis; hyperglycemia promotes glycation of endothelium and promotes oxidative stress
- Inflammation / CRP: High-sensitivity CRP is an independent predictor of risk; reflects the inflammatory milieu driving plaque progression
Pathogenesis
The process is driven by an interplay of vessel wall injury and chronic inflammation. The "response-to-injury" hypothesis underlies all modern understanding.
Step 1 - Endothelial Injury and Dysfunction
Any persistent or repetitive insult (hemodynamic stress, oxidized LDL, cigarette smoke toxins, hyperglycemia, immune mechanisms, infection) causes endothelial cell (EC) dysfunction. This:
- Increases expression of adhesion molecules (VCAM-1, ICAM-1, selectins)
- Decreases nitric oxide (NO) production, which normally prevents platelet adhesion and monocyte recruitment
- Increases permeability to lipoproteins
Step 2 - Lipid Accumulation
LDL particles accumulate in the intima. They undergo oxidation by reactive oxygen species (ROS) generated by macrophages and ECs. Oxidized LDL (oxLDL) is key - it is:
- Directly cytotoxic to endothelium
- Chemotactic for monocytes
- An activator of macrophage scavenger receptors
Step 3 - Monocyte Recruitment and Foam Cell Formation
Circulating monocytes adhere to activated endothelium via adhesion molecules, migrate into the intima, and differentiate into macrophages. These macrophages phagocytose oxLDL via scavenger receptors (not the regulated LDL receptor), leading to uncontrolled lipid accumulation. The result: macrophage foam cells - the hallmark early cell of atherosclerosis. Foam cells aggregate to form fatty streaks (the earliest visible lesion).
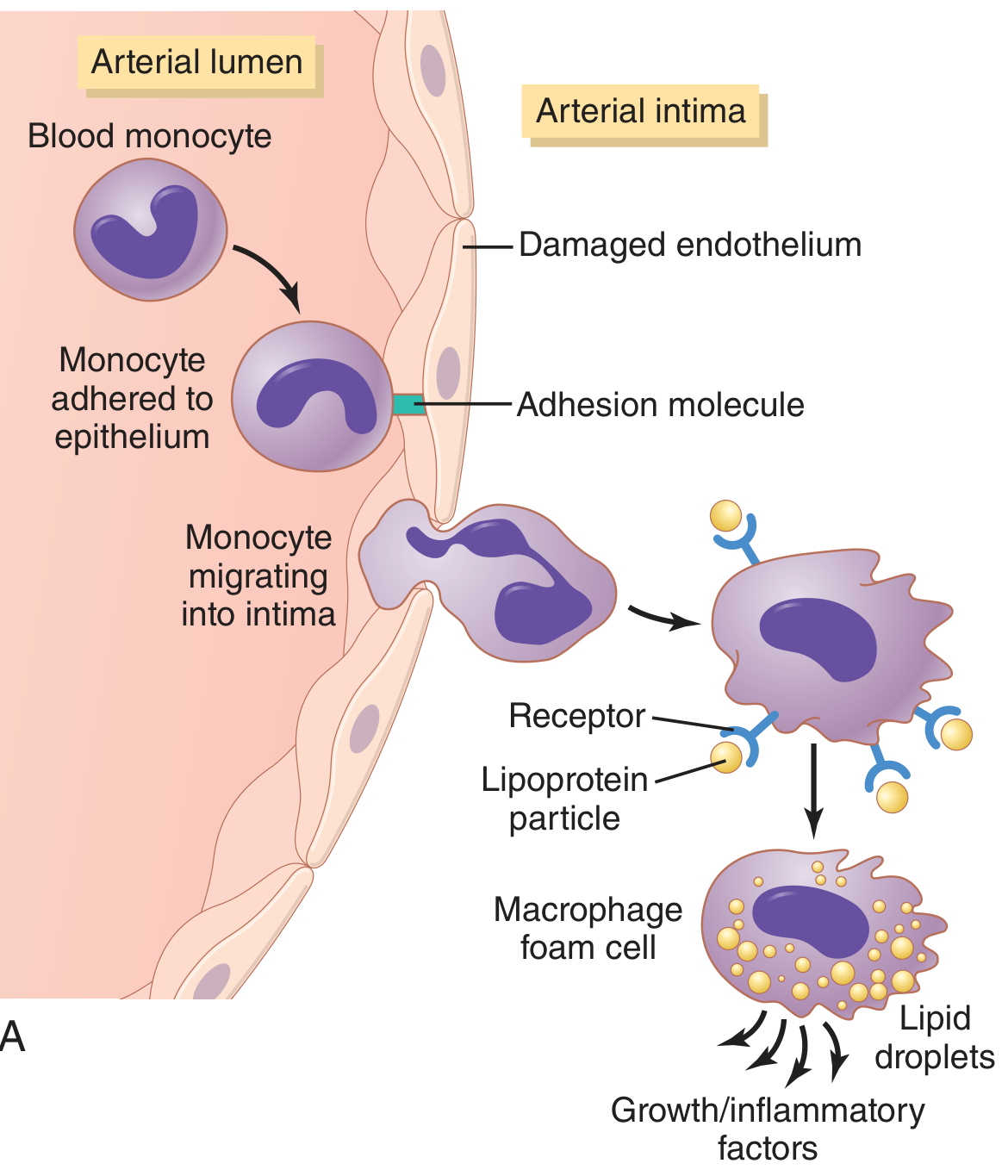
Step 4 - Smooth Muscle Cell (SMC) Proliferation and Matrix Synthesis
Growth factors released by platelets (PDGF), macrophages, and ECs recruit SMCs from the media into the intima (or from circulating precursors). Intimal SMCs:
- Proliferate
- Synthesize ECM (especially collagen), which forms the fibrous cap
- This converts a fatty streak into a mature atherosclerotic plaque
Fibroblast growth factor (FGF) and PDGF are major drivers of this step.
Step 5 - Plaque Growth and Complication
Plaques grow slowly over decades. Inflammatory cytokines (e.g., IFN-γ from T cells, TNF and IL-1 from macrophages) can inhibit SMC collagen synthesis and cause SMC apoptosis, thinning the cap and creating an unstable (vulnerable) plaque.
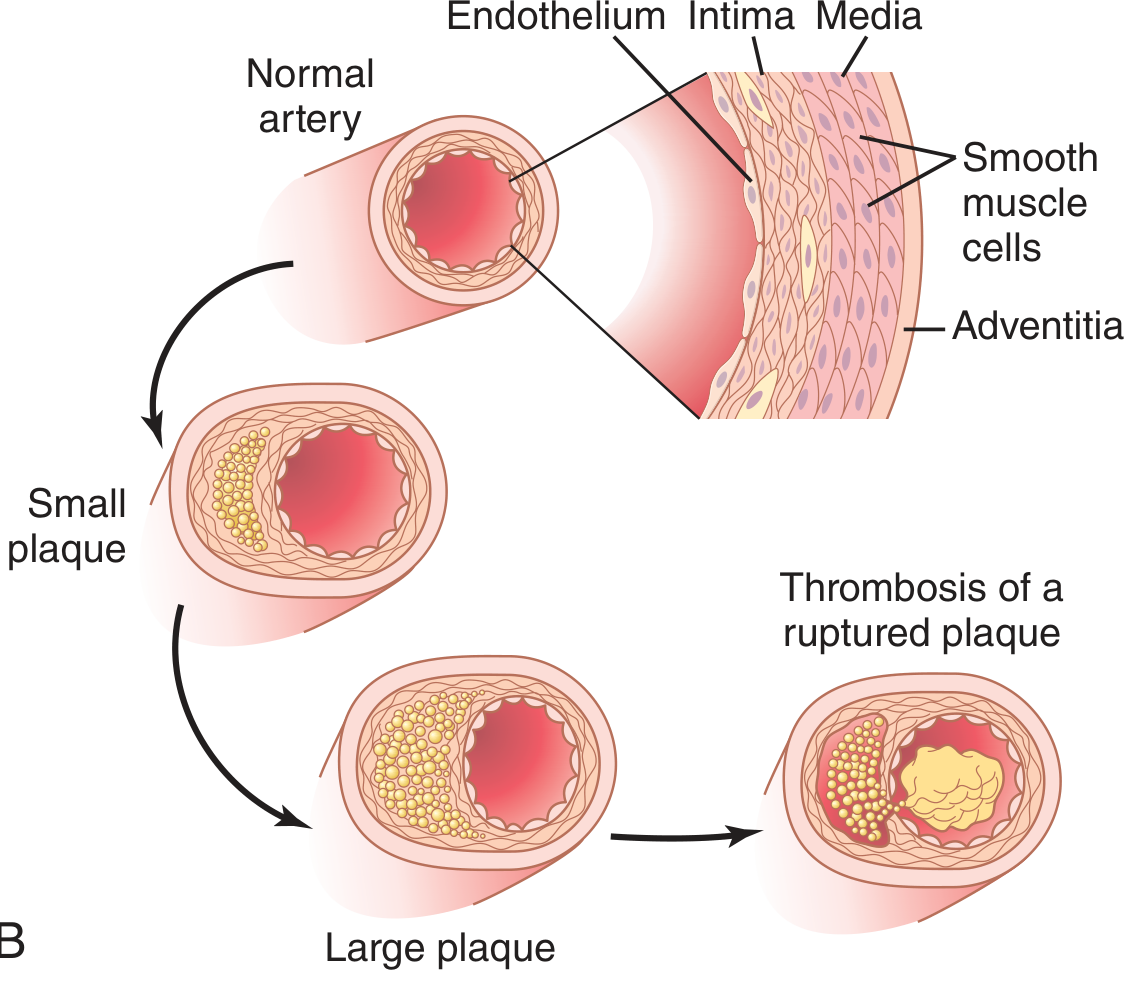
Morphology: Progression of Lesions
| Stage | Features |
|---|---|
| Fatty streak | Minute yellow, flat macules; lipid-filled foam macrophages only; appear in aortas of infants < 1 year; present in all children > 10 years; minimally raised, no flow disturbance |
| Atherosclerotic plaque | White-yellow raised intimal lesions, 0.3-1.5 cm; eccentric (patchy) involvement; fibrous cap + lipid core; range from stable to unstable |
| Complicated plaque | Calcification, ulceration, superimposed thrombosis (red-brown color), aneurysm formation |
Plaque composition:
- Fibrous cap: SMCs, dense collagen, proteoglycans, elastin fibers; overlying EC layer
- Lipid/necrotic core: Cholesterol, cholesterol esters, cellular debris, foam cells, calcification
- Periphery ("shoulder"): Active zone with macrophages, T cells, SMCs - the most vulnerable zone for rupture
Preferred sites (descending severity):
- Infrarenal abdominal aorta
- Coronary arteries
- Popliteal arteries
- Internal carotid arteries
- Circle of Willis
Stable vs. Vulnerable (Unstable) Plaques
| Feature | Stable Plaque | Vulnerable (Unstable) Plaque |
|---|---|---|
| Fibrous cap | Thick, dense collagen | Thin, degraded by MMPs |
| Lipid core | Small | Large, soft |
| Inflammation | Minimal | Dense macrophage/T-cell infiltrate |
| Calcification | Often present | Less prominent |
| Risk | Chronic stenosis, stable angina | Plaque rupture → ACS, MI, stroke |
Vulnerable plaques rupture when metalloproteinases (MMPs) secreted by activated macrophages degrade the fibrous cap collagen. Rupture exposes the thrombogenic lipid core to blood, triggering platelet aggregation and thrombus formation - the cause of most acute coronary syndromes.
Clinical Consequences
Atherosclerosis underlies virtually all major cardiovascular events:
- Coronary artery disease (CAD): Stable angina (chronic stenosis), unstable angina, myocardial infarction (plaque rupture + thrombosis)
- Cerebrovascular disease: Ischemic stroke, TIA (carotid and intracranial atherosclerosis)
- Peripheral arterial disease (PAD): Claudication, critical limb ischemia, gangrene
- Aortic aneurysm: Medial ischemia from atherosclerosis weakens the aortic wall
- Renovascular hypertension: Renal artery stenosis from plaques
Key Cells and Mediators Summary
| Cell/Molecule | Role |
|---|---|
| Endothelial cells | First responders to injury; express adhesion molecules; reduce NO |
| Monocytes/Macrophages | Phagocytose oxLDL → foam cells; release growth factors, MMPs, cytokines |
| Smooth muscle cells | Migrate from media; proliferate; synthesize collagen (cap formation) |
| T lymphocytes | Secrete IFN-γ; promote inflammation; inhibit SMC repair |
| Oxidized LDL | Toxic to endothelium; drives foam cell formation via scavenger receptors |
| PDGF, FGF | Drive SMC migration and proliferation |
| MMPs (matrix metalloproteinases) | Degrade fibrous cap; key mediators of plaque instability |
| NO (nitric oxide) | Vasodilatory; anti-adhesive; reduced in atherosclerosis |
Prevention and Treatment Principles
- Statins: Reduce LDL synthesis; also have pleiotropic anti-inflammatory effects on plaques
- PCSK9 inhibitors: Further reduce LDL in high-risk patients
- Antihypertensives: Reduce hemodynamic shear stress
- Smoking cessation: Reverses endothelial injury, reduces pro-thrombotic state
- Antiplatelet therapy (aspirin, clopidogrel): Reduce thrombotic complications, especially post-ACS
- Glycemic control in DM: Slows the accelerated atherogenesis seen in diabetes
- Diet and exercise: Reduce multiple modifiable risk factors simultaneously
A 2025 Frontiers in Immunology review highlights emerging targets including metabolic reprogramming of immune cells, phenotypic switching of vascular SMCs, and novel forms of programmed cell death (e.g., pyroptosis, ferroptosis) as next-generation therapeutic directions.
Primary sources: Robbins & Kumar Basic Pathology (10th ed.), p. 313-321; Guyton and Hall Textbook of Medical Physiology, p. 850-852
This is a shared conversation. Sign in to Orris to start your own chat.