Epidermylosis bullosa
Reading File
Finding Sources
Searching PubMed
"epidermolysis bullosa"[MeSH Terms]
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Reading File
Reading File
Reading File
Searching the Web
epidermolysis bullosa treatment 2025 gene therapy FDA approved krystal biotech
Reading File
Searching Images
epidermolysis bullosa blistering skin lesions subtypes
Now I have comprehensive information to provide a thorough answer. Let me compile it.
Epidermolysis Bullosa (EB)
Overview
Epidermolysis bullosa is a family of inherited genodermatoses characterized by mechanical fragility of the skin resulting in blister formation in response to minor trauma. Patients develop small vesicles or larger bullae on cutaneous and, in severe forms, mucosal surfaces. The depth of blister formation, distribution, extracutaneous involvement, and severity vary with the specific subtype and underlying molecular defect.
— Fitzpatrick's Dermatology, Chapter 60
Classification: 4 Main Types
EB is classified by the level of skin cleavage (blister plane):
| Type | Cleavage Plane | Key Mutated Proteins |
|---|---|---|
| EB Simplex (EBS) | Intraepidermal | Keratins 5 & 14 (KRT5, KRT14) |
| Junctional EB (JEB) | Within lamina lucida | Laminin-332, Collagen XVII (BP180), Integrin α6β4 |
| Dystrophic EB (DEB) | Below lamina densa (uppermost dermis) | Collagen VII (COL7A1) |
| Kindler Syndrome | Mixed/variable | Kindlin-1 (FERMT1) |
1. EB Simplex (EBS)
Most commonly autosomal dominant; caused by keratin gene mutations. Blistering is intraepidermal and heals without scarring.
Subtypes:
- Generalized Severe (Dowling-Meara / Herpetiformis): Presents at birth; most severe EBS. Grouped "herpetiform" blisters on trunk/proximal extremities. Oral mucosa often involved. Nail shedding/dystrophy. Palmoplantar hyperkeratosis (keratoderma). May affect esophagus and larynx.
- Generalized Intermediate (Koebner): Generalized blistering from birth or early infancy; hands, feet, extremities most affected. Postinflammatory hypo/hyperpigmentation. Mild oral involvement that improves with age.
- Localized (Weber-Cockayne): Mild form; predominantly palmoplantar blistering triggered by friction/heat.
- Uncommon variants: EBS with muscular dystrophy (plectin mutations), EBS with mottled pigmentation, suprabasal EBS subtypes.
2. Junctional EB (JEB)
Autosomal recessive. Cleavage in the lamina lucida; blistering involves mucosae prominently.
Subtypes:
- Herlitz JEB (Lethal): Caused by null mutations in laminin-332 genes (LAMA3, LAMB3, LAMC2) → complete absence of laminin-332. Widespread blistering + exuberant granulation tissue around mouth/nose/tracheostomy. High neonatal/infantile mortality.
- Non-Herlitz JEB (GABEB — Generalized Atrophic Benign EB): Most caused by mutations in COL17A1 (Collagen XVII/BP180). Blistering in lamina lucida, with hemidesmosomal abnormalities; nail loss and skin atrophy. Better prognosis than Herlitz.
- JEB with pyloric atresia: Associated with ITGA6/ITGB4 mutations.
- Laryngo-Onycho-Cutaneous syndrome: N-terminal mutations of LAMA3.
Immunofluorescence tip: Absence of laminin α3 staining suggests an α3 gene mutation; ~80% of laminin-332 mutations involve recurrent nonsense mutations in LAMB3.
3. Dystrophic EB (DEB)
Caused by mutations in COL7A1 (collagen VII), a component of anchoring fibrils below the lamina densa. Blisters heal with scarring.
Subtypes:
- Dominant DEB: Typically localized; milder scarring. Nail dystrophy is common. Pretibial EB is a classic dominant variant.
- Recessive DEB (RDEB):
- Mild/Localized RDEB: Acral blistering; mild or absent mucosal involvement.
- Severe RDEB (Hallopeau-Siemens): Devastating multisystem disease. Presents with generalized blistering at birth, sometimes with congenital absence of skin (Bart syndrome). Progressive scarring → pseudosyndactyly ("mitten hand" deformity), flexion contractures. Oropharyngeal involvement leads to microstomia, esophageal strictures, dental caries, and malnutrition. Anemia (chronic inflammation + hemorrhage). Major complication: invasive squamous cell carcinoma (SCC) — leading cause of death in adults with severe RDEB.

Extracutaneous Complications (Severe Forms)
| System | Complication |
|---|---|
| GI | Esophageal strictures, webbing, pyloric atresia |
| Oral | Microstomia, enamel hypoplasia, dental caries |
| Eyes | Corneal erosions, scarring |
| Respiratory | Laryngeal/tracheal narrowing, need for tracheostomy |
| Hematologic | Chronic anemia |
| Nutritional | Malnutrition, growth retardation |
| Oncologic | Aggressive SCC (RDEB), melanoma |
| Nails | Onycholysis, nail loss, pterygium, periungual granulation tissue |
Diagnosis
- Clinical history & physical exam — blistering pattern, distribution, age of onset, family history
- Skin biopsy (perilesional skin):
- Transmission electron microscopy (TEM) — identifies cleavage plane precisely
- Immunofluorescence mapping (IFM) — antibodies against laminin-332, collagen XVII, collagen VII, integrin β4 to localize protein defects
- DNA mutational analysis — confirms diagnosis, guides prognosis, enables:
- Genetic counseling
- Prenatal diagnosis (chorionic villus sampling at 8–10 weeks, amniocentesis)
- Preimplantation genetic diagnosis
RDEB and dominant DEB may have equivalent blistering activity, but only RDEB carries high SCC risk — DNA diagnosis distinguishes them.
Treatment
Supportive (mainstay)
- Wound care: Soak in modified Dakin solution (0.025% sodium hypochlorite) 20 min before dressing changes to debride and reduce bacterial load; apply mupirocin or topical antibiotics; cover with non-adherent, semi-occlusive dressings; avoid tape (causes further blistering); use self-adhering gauze.
- Blister management: New blisters should be lanced with sterile needle to prevent extension; do not remove the blister roof.
- Infection control: S. aureus and S. pyogenes are the most common pathogens. Systemic antibiotics for infected wounds.
- Nutritional support: High-calorie diet; gastrostomy tube placement when oral intake is severely compromised.
- Heat avoidance (especially for EBS): Soft, well-ventilated footwear.
- Hand surgery: For pseudosyndactyly in RDEB; post-op splinting essential to prevent recurrence.
Multidisciplinary care
Involves dermatology, surgery, gastroenterology (esophageal dilatations), dentistry, ophthalmology, nutrition, and anesthesia. Anesthetic challenges include friable mucosa — airway management requires extreme caution.
Emerging/Disease-Modifying Therapies
- Gene therapy: Cell-based gene correction for RDEB (COL7A1-corrected keratinocyte grafts; iPS cell approaches using CRISPR/Cas9).
- Beremagene geperpavec (B-VEC / Vyjuvek): FDA-approved 2023 — topical HSV-1 vector delivering COL7A1 gene directly to wounds in RDEB. First approved gene therapy for EB.
- Protein replacement therapy: Recombinant collagen VII infusions.
- Stem cell transplantation: Bone marrow/allogeneic HSCT in RDEB — trials show partial improvement.
- Angiotensin receptor antagonists: Losartan has shown benefit in RDEB (reduces TGF-β-driven fibrosis). A 2025 systematic review (PMID: 40898775) assessed the safety and efficacy of ARBs in RDEB.
Surveillance & Complications Monitoring (RDEB)
- Regular skin exam for SCC — aggressive behavior, early metastasis; screening from adolescence onward
- CBC for anemia, serum ferritin, albumin, pre-albumin (nutritional markers)
- Barium swallow / endoscopy for esophageal involvement
- Ophthalmologic exams
Clinical Images
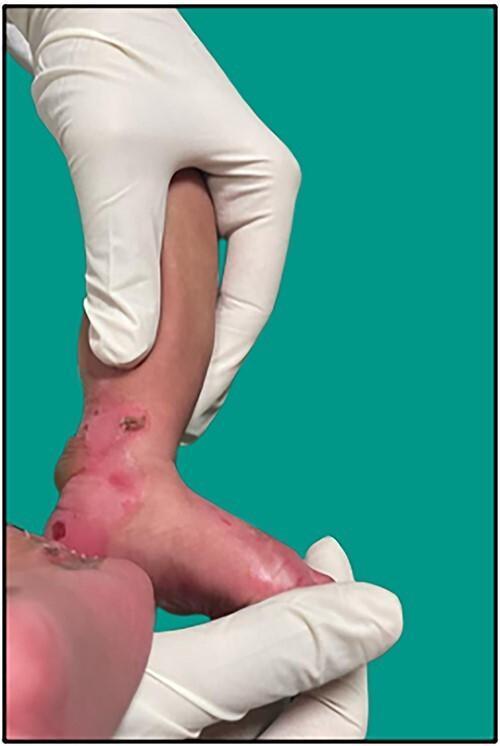
Neonatal lower limb showing ruptured bullae, erosions, and crusting at trauma-prone sites — classic presentation of EB in the newborn.
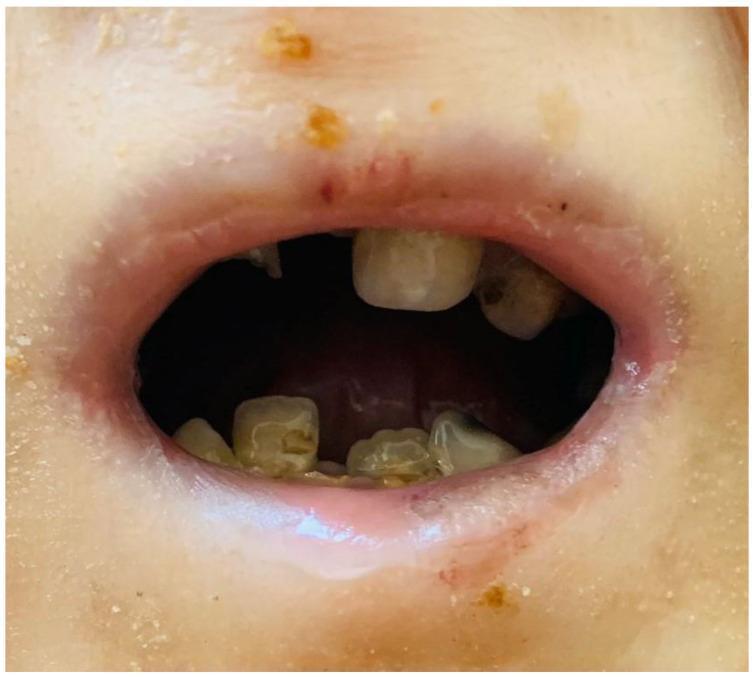
Perioral scarring, microstomia, and severe enamel hypoplasia/caries in a patient with dystrophic/junctional EB.
Sources: Fitzpatrick's Dermatology (Chapter 60 — Inherited EB); Dermatology 2-Volume Set 5e (Chapters 32, 95); Fitzpatrick's Neonatology section; RDEB SCC surveillance data from Fitzpatrick's block 15.
This is a shared conversation. Sign in to Orris to start your own chat.