Explain the mechanism of acute respiratory distress syndrome
Reading File
Finding Sources
Finding Sources
Searching PubMed
"acute respiratory distress syndrome"[MeSH Terms] AND pathophysiology
Reading File
Reading File
Reading File
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Reading File
I now have all the information needed for a comprehensive, well-illustrated answer. Let me compile this:
Acute Respiratory Distress Syndrome (ARDS): Mechanism
ARDS is a syndrome of non-cardiogenic pulmonary edema caused by diffuse alveolar injury, triggering a cascade of inflammatory, cellular, and physiological events that progressively impair gas exchange. Its natural history is divided into three overlapping phases.
Triggers and Initial Injury
ARDS arises from both direct and indirect lung injury:
| Direct Lung Injury | Indirect Lung Injury |
|---|---|
| Pneumonia | Sepsis |
| Gastric aspiration | Severe trauma / multiple fractures |
| Pulmonary contusion | Burns |
| Near-drowning | Multiple transfusions |
| Toxic inhalation | Drug overdose / pancreatitis |
More than 80% of cases result from pneumonia, sepsis, aspiration, or trauma. The common endpoint is activation of innate immunity and a destructive inflammatory cascade within the alveoli.
- Harrison's Principles of Internal Medicine 22E, p. 2343
Phase 1: Exudative Phase (Days 0-7)
This is the acute inflammatory phase, characterized by breakdown of the alveolar-capillary barrier.
Cellular and molecular events
-
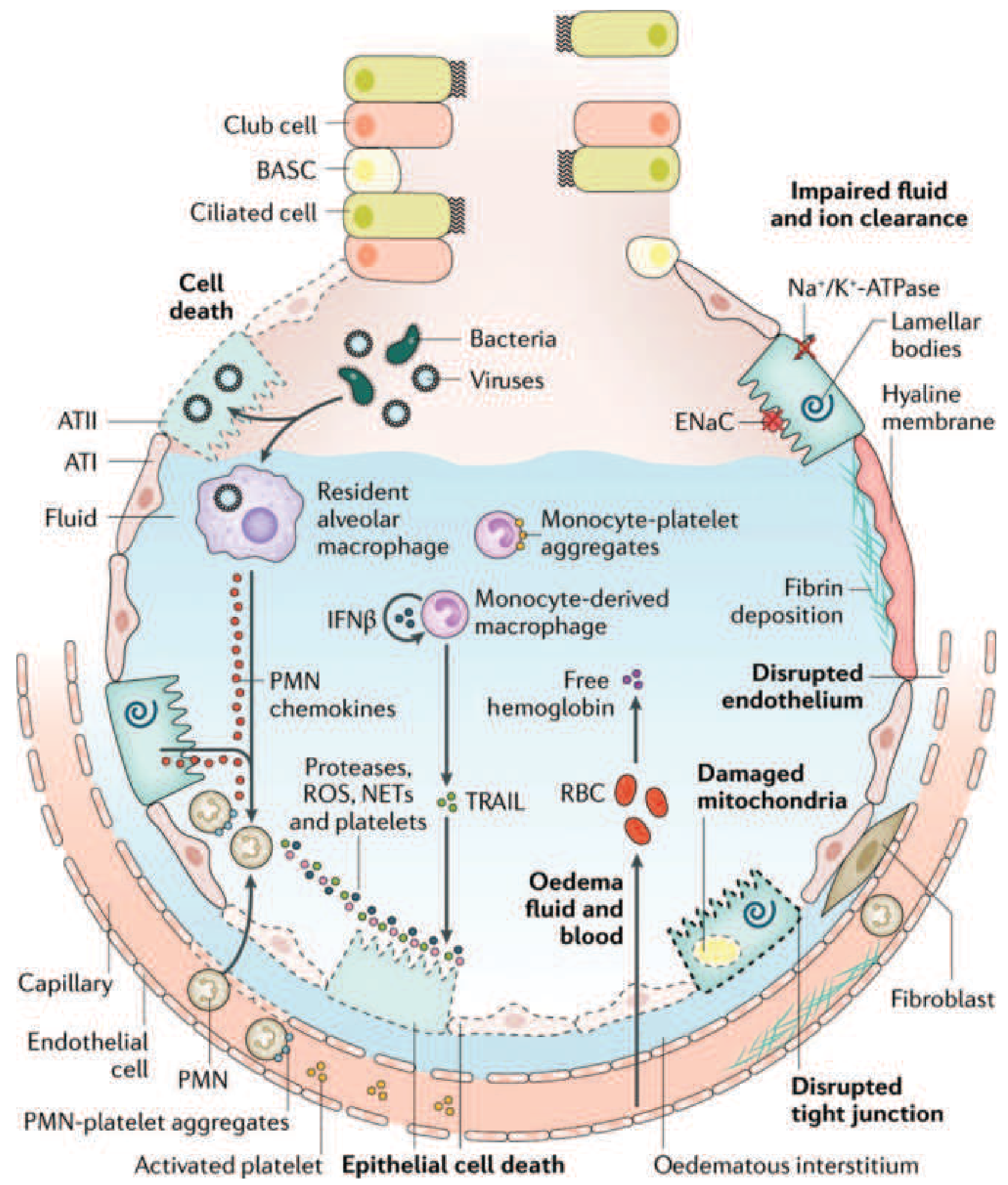
Initial insult activates Toll-like receptors on type I alveolar epithelial cells (ATI) and resident alveolar macrophages, triggering release of proinflammatory chemokines (IL-1, IL-6, IL-8, TNF-α, leukotriene B2).
-
Neutrophil recruitment: Chemokines recruit circulating polymorphonuclear (PMN) leukocytes into the pulmonary interstitium and alveolar space. Activated neutrophils release:
- Proteases (elastase, metalloproteinases) - degrade the basement membrane
- Reactive oxygen species (ROS) - cause direct oxidative injury
- Neutrophil extracellular traps (NETs) - trap pathogens but also damage epithelium
-
Monocyte and platelet involvement: Monocytes migrate into the lung and cause epithelial cell apoptosis via IFN-β-dependent release of TRAIL (TNF-related apoptosis-inducing ligand). Activated platelets form aggregates with PMNs and monocytes, amplifying injury. Red blood cells release cell-free hemoglobin, further exacerbating oxidative damage.
-
Endothelial and epithelial barrier failure: Endothelial tight junctions widen and intercellular junctions disrupt. Type I pneumocytes undergo necrosis. The result is massive protein-rich edema fluid flooding the interstitium and alveolar spaces.
-
Surfactant dysfunction: Alveolar edema washes out and inactivates surfactant, causing alveolar collapse (atelectasis) and a dramatic reduction in lung compliance. Phospholipase A2 (elevated in pancreatitis-related ARDS) directly degrades surfactant phospholipids.
-
Hyaline membrane formation: Condensed plasma proteins, fibrin, and cellular debris aggregate in the air spaces to form hyaline membrane whorls - the histologic hallmark of diffuse alveolar damage (DAD).
-
Vascular injury: Microthrombi and fibrocellular proliferation occlude the pulmonary microcirculation, causing pulmonary hypertension and increasing dead space.
Physiological consequences of the exudative phase
- Refractory hypoxemia: Large right-to-left intrapulmonary shunt forms because flooded/collapsed alveoli are still perfused. Mixed venous blood passes through these units without being oxygenated. Crucially, this shunt does not respond to supplemental oxygen alone - the hallmark of ARDS.
- Increased dead space: Microvascular occlusion reduces blood flow to ventilated lung units, causing hypercapnia and elevated minute ventilation requirements.
- Reduced lung compliance: The heavy, edematous, atelectatic lung (behaves like a sponge, with gravity-dependent consolidation) becomes stiff, increasing the work of breathing and leading to respiratory fatigue.
- Pulmonary hypertension: Results from hypoxic pulmonary vasoconstriction plus microvascular occlusion by microthrombi.
Clinical onset is typically within 12-36 hours of the precipitating insult (range up to 7 days). The patient develops tachypnea, dyspnea, and progressive hypoxemia requiring mechanical ventilation.
- Harrison's Principles of Internal Medicine 22E, pp. 2343-2344
- Fishman's Pulmonary Diseases and Disorders, pp. 2494-2495
Phase 2: Proliferative Phase (Days 7-21)
Many patients begin recovering during this phase, but others deteriorate:
- Type II pneumocyte proliferation along alveolar basement membranes begins repair - these cells synthesize new surfactant and differentiate into type I pneumocytes.
- A shift from neutrophil- to lymphocyte-predominant infiltrate occurs.
- Alveolar exudates start to organize.
- Fibroblast and myofibroblast proliferation begins, with early interstitial fibrosis in some patients.
- Dead space fraction and pulmonary hypertension may worsen as fibroproliferation remodels the vasculature.
Despite improving gas exchange, patients may still have significant hypoxemia and high minute ventilation requirements.
- Harrison's, p. 2344; Fishman's, p. 2495
Phase 3: Fibrotic Phase (Day 21 onwards - subset of patients)
Not all patients enter this phase, but those who do face significant morbidity:
-
Alveolar edema and inflammatory exudate convert to alveolar-duct and interstitial fibrosis.
-
Acinar architecture is destroyed, producing emphysema-like bullae.
-
Intimal fibroproliferation in the pulmonary microcirculation causes progressive vascular occlusion and worsening pulmonary hypertension.
-
Physiological consequences: further reduced compliance, increased dead space, risk of pneumothorax, and oxygen dependency.
-
Lung biopsy evidence of fibrosis in any ARDS phase is associated with increased mortality.
-
Harrison's, p. 2344-2345
Phase timeline summary

Key Pathophysiological Summary
| Mechanism | Effect |
|---|---|
| Barrier disruption (ATI + endothelium) | Protein-rich alveolar edema |
| Surfactant loss/inactivation | Atelectasis, reduced compliance |
| Neutrophil activation (ROS, proteases, NETs) | Direct cell injury, amplified inflammation |
| Cytokine storm (IL-1, IL-6, IL-8, TNF-α) | Sustained neutrophil recruitment |
| Microvascular occlusion | Dead space, pulmonary hypertension |
| Right-to-left shunt | Refractory hypoxemia |
| Fibroproliferation (late) | Irreversible structural remodeling |
The Berlin definition captures the clinical severity: PaO2/FiO2 ratio <300 mmHg (mild), <200 (moderate), <100 (severe), with bilateral opacities on imaging, onset within 7 days of a known insult, and no evidence that heart failure or fluid overload is the primary cause.
- Harrison's Principles of Internal Medicine 22E, p. 2343
- Fishman's Pulmonary Diseases and Disorders, pp. 2494-2495
- Sabiston Textbook of Surgery, p. 940
This is a shared conversation. Sign in to Orris to start your own chat.