I now have all the information needed to write a comprehensive, exam-mastery level learning note. Let me compile it now:
ANTIARRHYTHMICS: COMPLETE LEARNING NOTE
From Absolute Beginner to MBBS Examination Mastery
Sources: Goodman & Gilman's Pharmacological Basis of Therapeutics | Katzung's Basic & Clinical Pharmacology 16e | Lippincott Illustrated Reviews: Pharmacology | Harrison's Principles of Internal Medicine 22e | Braunwald's Heart Disease | Goldman-Cecil Medicine | Washington Manual of Medical Therapeutics
SECTION 1: BIG PICTURE OVERVIEW
What Problem Do Antiarrhythmic Drugs Solve?
Imagine your heart as a perfectly coordinated drum band. Every drummer (cardiac cell) plays at exactly the right time, producing a beautiful rhythm. The conductor (the SA node - sinoatrial node) sets the beat for everyone else. Every cell waits for the conductor's signal, fires in perfect sequence, and your heart beats regularly, pumping blood to the body.
Now imagine what happens if one random drummer in the group starts beating on their own - too fast, too early, or completely out of sync. Or imagine a signal getting "trapped" in a loop, causing the drums to keep firing in circles. The whole rhythm falls apart. The heart either beats too fast, too slow, or in a completely chaotic, disorganized pattern.
That is what an arrhythmia is: a disorder of the normal electrical rhythm of the heart.
When the rhythm goes wrong:
- The heart may beat too fast (tachyarrhythmia) and not fill with enough blood
- The heart may beat too slow (bradyarrhythmia) and not pump enough blood
- The ventricles may fibrillate (quiver uselessly) - this causes sudden cardiac death within minutes
- Blood may pool and clot (especially in atrial fibrillation), causing strokes
Antiarrhythmic drugs restore normal rhythm by controlling the electrical behavior of cardiac cells.
They do this by targeting the ion channels (tiny gates in the cell membrane) that create the electrical signals, or by targeting the nervous system's control over the heart.
SECTION 2: BUILD THE FOUNDATION
2A. Normal Cardiac Electrophysiology - Start From Scratch
Step 1: What IS electricity in the heart?
Your heart cells are surrounded by a thin wall called the cell membrane. On one side of this membrane are positively charged particles (ions) like sodium (Na+), potassium (K+), and calcium (Ca2+). On the other side are different concentrations of the same particles.
Think of it like a battery: when positive charges are separated from each other by a membrane, there is a difference in electrical charge - this is called the membrane potential (measured in millivolts, mV).
At rest, the inside of a cardiac cell is approximately -85 to -90 mV compared to the outside. This means the inside is negatively charged. This is called the resting membrane potential.
How does it stay negative? Because:
- A pump (Na+/K+-ATPase) actively pumps 3 Na+ OUT and only 2 K+ IN per cycle, creating a negative interior
- K+ channels are open at rest, letting K+ slowly leak OUT, making the inside even more negative
This is the "battery being charged" - the cell is ready to fire.
Step 2: The Action Potential - The Electrical Signal
When a cardiac cell receives a stimulus (from the cell next to it), it fires an action potential - a brief, rapid change in membrane potential that travels like a wave through the heart.
This is exactly like falling dominoes: one tile falls (fires), which knocks over the next one, and the signal travels from the SA node all the way through the heart to make it contract.
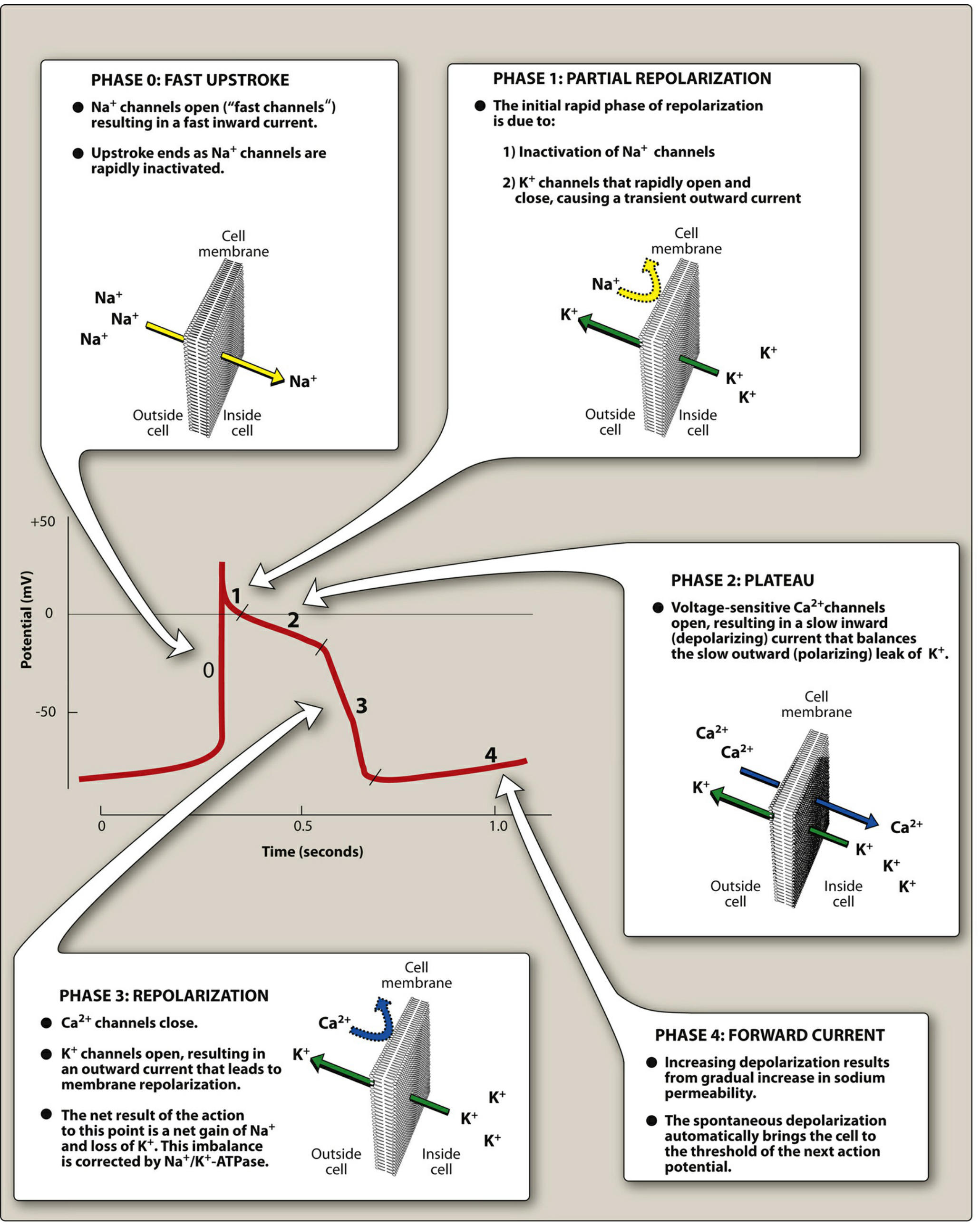
The action potential has 5 phases, each controlled by different ion channels:
Figure: The cardiac action potential with each phase and the corresponding ion movements. This diagram is central to understanding ALL antiarrhythmic drug mechanisms. - Lippincott Illustrated Reviews: Pharmacology
Let's go through each phase carefully:
PHASE 0 - The Fast Upstroke (Rapid Depolarization)
- What happens: Voltage-gated Na+ channels OPEN suddenly
- Massive Na+ rushes INTO the cell (Na+ loves to enter because the inside is very negative and the concentration is much higher outside)
- The inside of the cell shoots from -90 mV to +30 mV very rapidly
- Analogy: Like a dam bursting - water (Na+) rushes through the flood gates
Why this matters for drugs: Class I antiarrhythmics block Na+ channels and slow down Phase 0. This slows the speed of signal conduction.
PHASE 1 - Brief Initial Repolarization
- Na+ channels quickly INACTIVATE (close)
- A transient K+ current flows briefly outward
- The membrane voltage falls slightly from +30 mV back toward 0 mV
This phase is short and mostly relevant to Class IB drugs (like lidocaine) which shorten this phase.
PHASE 2 - The Plateau (The Most Unique Feature of Cardiac Cells)
- Voltage-gated Ca2+ channels (L-type) OPEN and Ca2+ flows INTO the cell
- This inward Ca2+ current is balanced by a slow outward K+ current
- The voltage "plateaus" near 0 mV for 200-300 milliseconds
- This Ca2+ is what triggers muscle contraction (excitation-contraction coupling)
- The plateau also creates the refractory period - the heart cannot be stimulated again until Phase 2 is over
Why this matters: The plateau (Phase 2) creates the refractory period. No matter how many signals arrive, the heart cannot fire again until it fully repolarizes. This prevents the heart from having "tetanic" (sustained) contractions like skeletal muscle.
Class IV drugs (Ca2+ channel blockers) act mainly at the SA and AV nodes, which are more dependent on Ca2+ for their action potentials.
PHASE 3 - Rapid Repolarization
- Ca2+ channels CLOSE
- K+ channels OPEN widely, letting K+ rush OUT
- The cell rapidly returns to negative resting potential (-85 to -90 mV)
- This is restoration of the "charged battery"
Why this matters for drugs: Class III antiarrhythmics BLOCK K+ channels. This prevents K+ from rushing out, prolonging Phase 3. This makes the action potential duration (APD) and the refractory period longer, preventing rapid re-firing.
PHASE 4 - Resting Membrane Potential (or Spontaneous Depolarization in Pacemaker Cells)
- In ordinary cardiac myocytes (working muscle cells): Phase 4 is flat - the cell just sits quietly at -90 mV waiting for the next signal
- In pacemaker cells (SA node, AV node, His-Purkinje system): Phase 4 is NOT flat - there is a slow, spontaneous inward "funny current" (If) that gradually depolarizes the cell toward threshold
- This is called automaticity - the ability to self-generate electrical signals
The SA node fires fastest (60-100 times per minute), so it is the dominant pacemaker. AV node fires at 40-60/min. Purkinje fibers fire at 20-40/min. These slower pacemakers are normally "overridden" by the SA node, but can take over if the SA node fails.
Why this matters: Class II drugs (beta-blockers) inhibit the sympathetic nerves that speed up Phase 4 depolarization in the SA and AV nodes. This is why beta-blockers slow the heart rate.
Key Difference: SA Node vs. Ventricular Muscle Cell Action Potential
The SA node (and AV node) are "slow response" cells that use Ca2+ channels predominantly (not Na+ channels) for their action potential upstroke. This is WHY:
- Class IV drugs (Ca2+ channel blockers like verapamil, diltiazem) preferentially affect the SA and AV nodes
- Class I drugs (Na+ channel blockers) mainly affect the atria, ventricles, and His-Purkinje system
SA NODE ACTION POTENTIAL: VENTRICULAR ACTION POTENTIAL:
No fast upstroke Fast upstroke (Phase 0) - Na+
Slow Phase 4 depolarization Flat Phase 4
Ca2+ driven Na+ driven initially, Ca2+ sustains plateau
Sensitive to Class II, IV Sensitive to Class I, III
2B. The Cardiac Conduction System - The Electrical Highway
SA NODE (Right atrium)
|
| → Interatrial pathways → LEFT ATRIUM
↓
AV NODE (junction of atria and ventricles)
↓ [Important delay here - ~0.12 sec - allows ventricles to fill]
BUNDLE OF HIS
↓
RIGHT BUNDLE BRANCH → Right ventricle
LEFT BUNDLE BRANCH → Left ventricle
↓
PURKINJE FIBERS (spread throughout ventricular walls)
↓
VENTRICULAR MYOCARDIUM contracts
Why the AV node delay matters: The 0.12-second pause at the AV node gives the atria time to finish contracting and push blood into the ventricles before the ventricles contract. This "top-up" is called the atrial kick. In atrial fibrillation, the atrial kick is lost.
Why this matters for drugs: Drugs that slow AV conduction (beta-blockers, Ca2+ channel blockers, digoxin, adenosine) are used to control ventricular rate in atrial arrhythmias, because they increase the AV nodal delay, meaning fewer atrial impulses reach the ventricles.
2C. How Arrhythmias Arise - The Two Main Mechanisms
Mechanism 1: Abnormal Automaticity
Normally, only the SA node fires spontaneously. But sometimes other cardiac cells develop the ability to fire on their own - this is called abnormal automaticity or ectopic automaticity.
Why does it happen?
- Myocardial ischemia (low oxygen) damages cells, making them fire erratically
- Hypokalemia (low K+) alters the resting membrane potential
- Sympathetic overstimulation speeds up Phase 4 depolarization everywhere
- Injury or stretching of cardiac tissue
Result: An ectopic pacemaker fires faster than the SA node and "takes over" the rhythm - like a rogue conductor grabbing the baton.
Analogy: Imagine a classroom where the teacher (SA node) normally sets the pace. If a student starts shouting instructions faster than the teacher, chaos ensues.
How drugs help: Class I and II drugs suppress abnormal automaticity by making Phase 4 depolarization slower or raising the threshold (the voltage the cell must reach before it can fire).
Mechanism 2: Re-entry (The Most Common Cause of Clinical Arrhythmias)
Re-entry is the single most important mechanism for clinical arrhythmias. It explains most SVTs, atrial flutter, atrial fibrillation, ventricular tachycardia, and WPW syndrome.
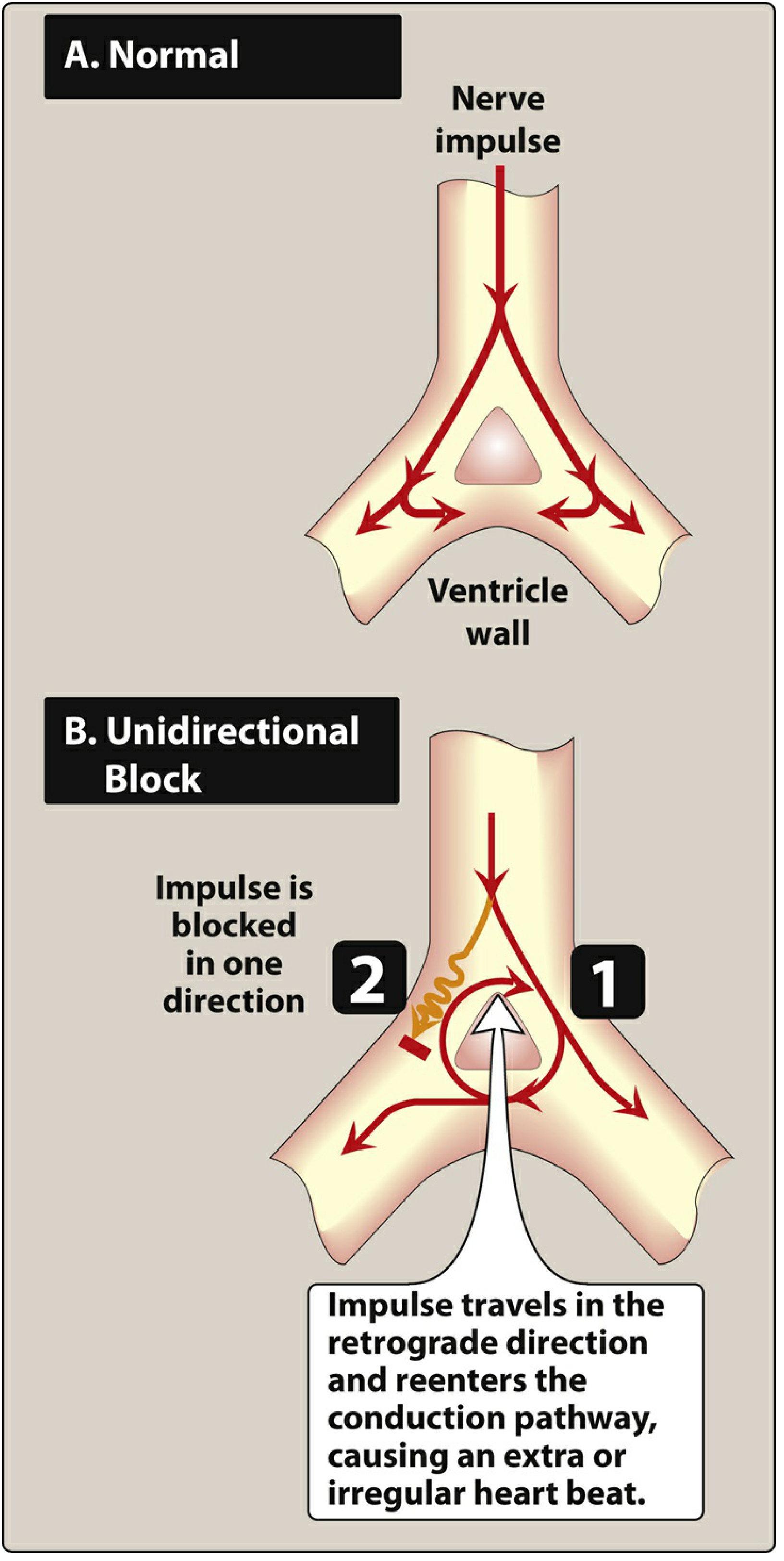
Figure: Schematic of reentry. In normal conduction (A), the impulse travels down two pathways and terminates. In unidirectional block (B), the impulse is blocked in one direction but travels retrogradely through the other, re-entering the circuit and causing continuous circular excitation. - Lippincott Illustrated Reviews: Pharmacology
What is re-entry? Step-by-step:
Imagine a road system where an electrical impulse travels like a car:
- Normally, the impulse travels down TWO parallel pathways (Path A and Path B)
- Both paths meet at the bottom, the impulse collides with itself and dies out
- Everything is normal. One beat. Over.
Now imagine Path A is blocked in one direction (unidirectional block) - perhaps due to ischemia:
- The impulse travels down Path B normally
- At the bottom, instead of dying, it turns backward through Path A (which allows backward travel, just not forward)
- It travels back up Path A, exits, and re-enters the circuit at the top
- It travels down Path B again...and again...and again
- The circuit keeps going - circular re-excitation producing continuous arrhythmia
For re-entry to occur, three things must coexist:
- Two pathways (anatomical or functional)
- Unidirectional block in one pathway (the impulse can only go one way)
- Slow conduction in the other pathway (so the first pathway has time to recover and allow retrograde conduction)
Conditions that cause re-entry:
- Myocardial infarction (scar tissue creates slow conduction zones)
- Hypertrophy
- Accessory pathways (WPW - Wolff-Parkinson-White syndrome, where an extra electrical connection between atria and ventricles exists)
- AV nodal re-entry (most common SVT)
How drugs terminate re-entry:
There are two approaches:
- Slow conduction further (Class I Na+ channel blockers) - make the unidirectional block into a bidirectional block, so the impulse cannot travel in ANY direction through the damaged area. Circuit breaks.
- Prolong the refractory period (Class III K+ channel blockers) - make cells stay refractory longer. When the circling impulse arrives, it finds the tissue still refractory (unable to respond). Circuit dies out.
Mechanism 3: Triggered Activity (Less Common but Important)
After a normal action potential, sometimes there are small "afterdepolarizations" - extra little bumps in voltage that can trigger another action potential.
Two types:
- Early Afterdepolarizations (EADs): Occur during Phase 2 or Phase 3. Caused by prolonged action potential duration (e.g., from hypokalemia, drugs that prolong QT). EADs are responsible for Torsades de Pointes (TdP) - a dangerous ventricular tachycardia.
- Delayed Afterdepolarizations (DADs): Occur after Phase 3, during Phase 4. Caused by intracellular Ca2+ overload (e.g., digitalis toxicity, catecholamine excess, heart failure). DADs are responsible for digitalis-induced arrhythmias.
2D. The ECG as a Window Into Electrical Activity
Every drug's effect on the ECG can be predicted from its mechanism:
PR INTERVAL: Time for impulse to go SA node → AV node → ventricles
Prolonged by: β-blockers, Ca²⁺ blockers, digoxin, adenosine
(They slow AV nodal conduction)
QRS COMPLEX: Time for ventricular depolarization (Phase 0 in ventricles)
Widened by: Class I drugs (Na⁺ channel blockers slow Phase 0)
QT INTERVAL: Total ventricular depolarization + repolarization time
Prolonged by: Class IA drugs (prolong Phase 3 also)
Class III drugs (block K⁺ channels, prolong Phase 3)
QT prolongation = RISK of Torsades de Pointes
High-yield ECG fact: QTc (corrected QT) > 500 ms = HIGH risk of Torsades de Pointes.
The corrected QT is calculated using Bazett's formula: QTc = QT / √RR
SECTION 3: DRUG CLASS FRAMEWORK
The Vaughan-Williams Classification
The Vaughan-Williams classification (1970) is the most widely used system for categorizing antiarrhythmic drugs. It groups them by their predominant effect on the cardiac action potential.
CLASS I → Na⁺ channel blockers
IA → Intermediate block, prolong APD
IB → Fast on/off, shorten APD
IC → Slow, marked conduction slowing
CLASS II → Beta-adrenergic blockers (β-blockers)
CLASS III→ K⁺ channel blockers (prolong APD/refractory period)
CLASS IV → Ca²⁺ channel blockers (non-dihydropyridine)
UNCLASSIFIED: Adenosine, Digoxin, Magnesium
Important limitation: Many drugs have actions in multiple classes. Amiodarone, for example, has Class I, II, III, and IV actions. Sotalol has Class II AND III actions. The Vaughan-Williams classification describes the predominant action, not the only one.
CLASS IA ANTIARRHYTHMICS
Quinidine, Procainamide, Disopyramide
Definition
Class IA agents are Na+ channel blockers with intermediate binding kinetics that also block K+ channels, producing intermediate slowing of Phase 0 AND prolongation of Phase 3.
Mechanism of Action - Detailed
Na+ channel blockade (Phase 0 effect):
- Na+ channels exist in three states: Open (during Phase 0), Inactivated (during phases 1-3), and Resting/Closed (during Phase 4)
- Class IA drugs bind to Na+ channels in their OPEN or INACTIVATED state
- They have intermediate kinetics - they bind and unbind at a moderate rate (this is "use-dependent" block: the faster the heart beats, the more channels are blocked)
- Blocking Na+ channels slows Phase 0 (the upstroke), which slows conduction velocity
- ECG effect: WIDENED QRS complex
K+ channel blockade (Phase 3 effect):
- Class IA drugs ALSO block some K+ channels (Ikr channels)
- This slows K+ efflux, prolongs Phase 3 repolarization
- The action potential duration (APD) and effective refractory period (ERP) are PROLONGED
- ECG effect: PROLONGED QT interval
This combined Na+ and K+ blockade is the hallmark of Class IA - they affect both conduction AND repolarization.
The Three Class IA Drugs - Individual Profiles
QUINIDINE (the original, the prototype)
Mechanism: Na+ channel block + K+ channel block + anticholinergic + alpha-blocking effects
Clinical Uses:
- Atrial fibrillation (AF) and atrial flutter - converting to sinus rhythm (largely replaced by newer drugs)
- Wolff-Parkinson-White (WPW) syndrome - can block the accessory pathway
- Historically: malaria treatment (quinidine is the dextro-rotatory isomer of quinine)
Adverse Effects (extremely high-yield - examiners love to test these):
-
Cinchonism: A syndrome caused by high quinidine levels. Symptoms: tinnitus (ringing in ears), headache, visual disturbances (blurred vision, photophobia), confusion, diarrhea. Named after the cinchona tree from which quinine (and quinidine) is derived.
-
Quinidine syncope: Paradoxically, quinidine can CAUSE arrhythmias (proarrhythmia). It prolongs the QT interval, which can trigger Torsades de Pointes (TdP) - a potentially fatal ventricular tachycardia. The patient suddenly loses consciousness (syncope).
-
Hypotension: Due to alpha-adrenergic blockade (vasodilation)
-
Diarrhea and GI upset: Very common, often the reason patients stop the drug
-
Thrombocytopenia and hemolytic anemia: Immune-mediated; rare
-
Drug interactions:
- Increases digoxin levels (VERY high-yield): Quinidine displaces digoxin from tissue-binding sites AND reduces digoxin's renal clearance. When starting quinidine in a patient on digoxin, the digoxin dose should be halved.
- Increases warfarin effects (inhibits CYP2C9): Increases bleeding risk
-
Anticholinergic effect (important): Quinidine also blocks muscarinic receptors. This can paradoxically SPEED UP AV nodal conduction (because normally the vagus nerve slows the AV node, and quinidine blocks this). In a patient with atrial flutter, this can dangerously accelerate the ventricular rate.
Contraindications: Pre-existing QT prolongation, heart failure (negative inotrope), severe conduction disease
PROCAINAMIDE
Mechanism: Na+ channel block. Its active metabolite, N-acetylprocainamide (NAPA), predominantly blocks K+ channels (Class III action).
Clinical Uses:
- WPW syndrome with AF - drug of choice in many guidelines (blocks both normal and accessory conduction)
- Acute ventricular tachycardia (IV form)
- Atrial fibrillation
- IV form useful in emergency settings
The Most Important Adverse Effect - Drug-Induced Lupus Erythematosus (DILE):
This is the single most tested fact about procainamide.
- With chronic use, procainamide causes a lupus-like syndrome in up to 25-30% of patients taking it long-term
- The acetylator status determines risk: Slow acetylators metabolize procainamide slowly, accumulate the drug, and are MORE prone to lupus
- Symptoms: arthralgia (joint pain), arthritis, pleuritis, pericarditis, skin rash
- ANA (antinuclear antibody) becomes positive in nearly ALL patients on long-term therapy
- Anti-histone antibodies are specifically associated (but the drug-induced form does NOT cause renal disease or CNS involvement unlike true SLE)
- Reversible on stopping the drug - this distinguishes it from true SLE
Other adverse effects:
- Agranulocytosis (decrease in white blood cells) - uncommon but dangerous
- QT prolongation and TdP (like all Class IA)
- Hypotension (IV administration)
- GI side effects
DISOPYRAMIDE
Mechanism: Na+ channel block + K+ channel block + most potent anticholinergic effect of all Class IA drugs
Clinical Uses:
- Atrial arrhythmias
- Hypertrophic obstructive cardiomyopathy (HOCM): The negative inotropic effect is actually useful here - it reduces the outflow tract obstruction. Disopyramide + beta-blocker is a well-known combination for HOCM.
Adverse Effects:
- Anticholinergic effects are predominant and the most tested:
- Urinary retention (especially in elderly men with BPH)
- Dry mouth
- Blurred vision
- Constipation
- Tachycardia
- Negative inotrope - can precipitate heart failure (most potent negative inotrope among Class I drugs)
- QT prolongation and TdP
- Contraindicated in: Glaucoma, BPH (urinary retention risk), pre-existing heart failure
Mnemonic: DISOPYRAMIDE = Dry mouth, Increase urinary retention, Slow heart failure patients, Often avoided
CLASS IA Summary Table
| Feature | Quinidine | Procainamide | Disopyramide |
|---|
| Na+ block | ++ | ++ | ++ |
| K+ block | ++ | + (via NAPA) | ++ |
| Anticholinergic | + | + | +++ (strongest) |
| Alpha block | + (hypotension) | - | - |
| Unique toxicity | Cinchonism, TdP, ↑digoxin | Drug-induced lupus (DILE) | Urinary retention, HF |
| Active metabolite | - | NAPA (Class III) | - |
| ECG effect | Wide QRS + Long QT | Wide QRS + Long QT | Wide QRS + Long QT |
CLASS IB ANTIARRHYTHMICS
Lidocaine, Mexiletine, Phenytoin
Definition
Class IB agents are Na+ channel blockers with rapid on-off kinetics (fast dissociation from channels) that preferentially affect ischemic or depolarized tissue, and they shorten the action potential duration.
Mechanism of Action - Detailed
Na+ channel blockade with rapid kinetics:
- Class IB drugs bind to Na+ channels in their OPEN or INACTIVATED state
- Key difference from Class IA: They dissociate (come off) the channels very RAPIDLY
- This means at normal heart rates, they barely affect healthy cells (the channels recover before the next beat)
- BUT in ischemic tissue where cells are partially depolarized (resting near the threshold), channels are more often in the INACTIVATED state
- Class IB drugs bind preferentially to INACTIVATED (partially depolarized) channels
- This is called "use-dependent" or "state-dependent" block - they work best on fast or ischemic tissue
Shortening of APD (Phase 3 effect):
- Unlike Class IA, Class IB drugs do NOT block K+ channels
- In fact, they actually slightly ENHANCE K+ current, shortening Phase 3
- This shortens the APD and effective refractory period
- ECG effect: NARROW QRS, QT may actually shorten (minimal ECG change)
Why this makes them useful for ventricular arrhythmias (not atrial):
- Ventricular cells have a longer APD than atrial cells
- Class IB drugs preferentially shorten the ventricular APD
- They also preferentially affect ischemic ventricular tissue (which is often the source of post-MI arrhythmias)
- They have NO significant effect on the SA or AV node
LIDOCAINE (IV only)
The prototype Class IB drug. Also the prototype local anesthetic.
Mechanism: Na+ channel blocker with rapid kinetics, preferential effect on depolarized (ischemic) tissue
Routes of Administration: INTRAVENOUS ONLY for antiarrhythmic use. NOT effective orally due to massive first-pass metabolism in the liver.
Clinical Uses:
- Acute ventricular arrhythmias - VT and VF post-MI (the classic indication)
- Ventricular arrhythmias during cardiac surgery or catheterization
- Digitalis-induced ventricular arrhythmias
- NOT effective for atrial arrhythmias (this is high-yield)
Pharmacokinetics:
- Half-life: about 2 hours
- Metabolized extensively by liver (CYP enzymes)
- In heart failure and liver disease, clearance is reduced - risk of toxicity
- Must be given as IV loading dose followed by continuous infusion
Adverse Effects - Almost entirely CNS:
The most testable fact: Lidocaine's toxicity is predominantly CNS (not cardiac).
Progressive CNS toxicity with increasing levels:
- Nystagmus - earliest sign (involuntary rapid eye movements)
- Dizziness, drowsiness
- Slurred speech, tinnitus
- Seizures - the most serious CNS effect
- Respiratory depression
- At very high levels: cardiac depression
Memory trick: "Lid's side effects LID the brain" - Nystagmus, then drowsiness, then speech problems, then seizures.
Drug interactions:
- Beta-blockers and cimetidine reduce lidocaine clearance → toxicity
- Phenytoin may accelerate lidocaine metabolism
MEXILETINE (Oral analogue of lidocaine)
Mechanism: Same as lidocaine - oral Class IB Na+ channel blocker
Clinical Uses:
- Oral suppression of ventricular arrhythmias (serves the same role as lidocaine but can be given by mouth)
- Also used in channelopathies (sodium channel disorders causing long QT syndrome type 3)
Adverse Effects:
- GI effects most common: Nausea, vomiting, dyspepsia, dysphagia (often taken with food)
- CNS effects similar to lidocaine: tremor, dizziness, coordination problems
- Narrow therapeutic index - need careful dosing
- Metabolized by CYP2D6 (important drug interaction with CYP2D6 inhibitors)
PHENYTOIN (Dilantin)
- Primarily an antiepileptic
- Also a Class IB-like antiarrhythmic
- Special use: Digitalis-induced arrhythmias (has been used historically)
- The pharmacology student should recognize this overlap between antiepileptic and antiarrhythmic actions
Class IB Key Points
| Feature | Lidocaine | Mexiletine |
|---|
| Route | IV only | Oral |
| Kinetics | Very fast on/off | Very fast on/off |
| Effect on APD | Shortens | Shortens |
| ECG effect | Minimal | Minimal |
| Main use | Acute VT/VF | Chronic ventricular arrhythmias |
| Main toxicity | CNS (seizures) | GI (nausea, vomiting) |
| Atrial arrhythmias | NOT effective | NOT effective |
CLASS IC ANTIARRHYTHMICS
Flecainide, Propafenone
Definition
Class IC agents are the most potent Na+ channel blockers. They have slow dissociation kinetics (they stay on the channel for a long time) and cause marked slowing of conduction without significantly affecting the action potential duration.
Mechanism of Action - Detailed
Na+ channel blockade - maximum potency:
- Class IC drugs bind Na+ channels very tightly (slow dissociation)
- They profoundly slow Phase 0 upstroke
- This markedly slows conduction velocity throughout the heart (especially in His-Purkinje and ventricular tissue)
- ECG effect: Markedly WIDENED QRS (sometimes very wide)
- They also slightly prolong the QT interval (due to QRS widening, not true K+ block)
- They do NOT significantly affect the action potential duration or refractory period
Why are they so dangerous in structural heart disease?
This is the most critical clinical point about Class IC drugs, and it is heavily tested.
The CAST Trial (Cardiac Arrhythmia Suppression Trial, 1989) was a landmark trial that shook the antiarrhythmic drug world:
- Patients after MI had frequent ventricular ectopic beats
- It was thought (logically) that suppressing these ectopic beats with antiarrhythmics would reduce mortality
- Flecainide and encainide (another Class IC drug) were used
- Result: MORTALITY INCREASED in the treatment group compared to placebo
- Flecainide and encainide INCREASED sudden cardiac death
Why? Because in ischemic/scarred tissue, marked conduction slowing by Class IC drugs creates NEW re-entry circuits and PROMOTES arrhythmias (proarrhythmic effect). The very drug intended to prevent arrhythmias was causing fatal ones.
The lesson from CAST: Class IC drugs are contraindicated in patients with structural heart disease (coronary artery disease, heart failure, left ventricular hypertrophy, prior MI).
FLECAINIDE
Clinical Uses:
- Atrial fibrillation and atrial flutter in patients with NO structural heart disease (the "Pill-in-the-pocket" approach - patient takes flecainide only when AF starts)
- Paroxysmal SVT
- WPW syndrome (with caution)
Adverse Effects:
- Proarrhythmia - most dangerous; CONTRAINDICATED in structural heart disease
- Visual disturbances (blurred vision, halos)
- Dizziness, headache
- Negative inotrope - avoid in heart failure
- Can convert AF to atrial flutter with very fast ventricular rate (flutter with 1:1 conduction - this is dangerous)
Contraindications: Structural heart disease (especially post-MI and heart failure), pre-existing bundle branch block
PROPAFENONE
Mechanism: Na+ channel block (Class IC) + weak beta-blocking activity + weak Ca2+ channel blocking activity
- The beta-blocking activity makes it slightly different from pure flecainide
- Can cause bronchospasm (due to beta-blockade) - avoid in asthma
- Metabolized by CYP2D6 (poor metabolizers have more beta-blocking effect)
Clinical Uses: Same as flecainide - AF and SVT in structurally normal hearts
Adverse Effects:
- Similar to flecainide
- Additional: Bronchospasm (beta-blockade), metallic taste, liver toxicity, agranulocytosis (rare)
- Increases digoxin and warfarin levels
Contraindications: Same as flecainide PLUS asthma/COPD (due to beta-blockade)
Class I Summary - The Big Picture
Class IA Class IB Class IC
Phase 0 Slow Slight slow Marked slow
Phase 3 Prolonged SHORTENED Unchanged
APD INCREASED Decreased Unchanged/slight increase
QRS Widened Minimal change MARKEDLY widened
QT Prolonged Unchanged/shorter Slightly prolonged (from QRS widening)
Best for Atrial + Ventricle Ventricle only Atrial (NO structural disease)
Ischemic Less selective Preferential Dangerous (proarrhythmia)
tissue
CLASS II ANTIARRHYTHMICS
Beta-Adrenergic Blockers (Beta-Blockers)
Definition
Class II antiarrhythmics are drugs that block beta-adrenergic receptors in the heart, reducing the influence of the sympathetic nervous system on cardiac automaticity and conduction.
Background Physiology
The sympathetic nervous system releases noradrenaline (norepinephrine) which binds to beta-1 receptors in the heart. This:
- Increases heart rate (chronotropy) - speeds up Phase 4 depolarization in SA node
- Increases AV nodal conduction speed (dromotropy) - shortens PR interval
- Increases the slope of Phase 4 in all pacemaker cells
- Increases contractility (inotropy)
Beta-blockers do the OPPOSITE:
- Slow Phase 4 depolarization in SA node → decreased heart rate
- Slow AV nodal conduction → prolonged PR interval, reduced ventricular rate
Mechanism of Action
Beta-blockers (propranolol, metoprolol, atenolol, esmolol) competitively block beta-1 receptors in the SA and AV nodes:
- SA node: Phase 4 depolarization slope decreases → heart rate decreases (bradycardia)
- AV node: Conduction velocity decreases → PR interval prolongs → fewer atrial impulses reach the ventricles
ECG effects: Bradycardia + prolonged PR interval
No significant effect on ventricular conduction or QRS width or QT interval (compared to Class I or III)
Clinical Uses (very high-yield)
- Post-MI arrhythmia prevention - THE drug class of choice post-MI. Beta-blockers reduce sudden cardiac death after MI by 20-25%.
- Rate control in atrial fibrillation and flutter - slow the ventricular response rate by blocking AV node
- Supraventricular tachycardias (SVT) - especially AV nodal re-entrant tachycardia (AVNRT)
- Sinus tachycardia due to sympathetic overactivity (thyrotoxicosis, anxiety, pheochromocytoma)
- Ventricular arrhythmias due to sympathetic excess (exercise-induced VT, congenital long QT syndrome type 1)
- Heart failure with reduced EF (carvedilol, metoprolol, bisoprolol) - reduce sudden death
Key Beta-Blocker Drugs in Antiarrhythmics
| Drug | Selectivity | Half-life | Route | Special Feature |
|---|
| Propranolol | Non-selective (β1 + β2) | 4-6 hours | Oral, IV | Prototype, lipophilic (CNS effects) |
| Metoprolol | Cardioselective (β1) | 3-7 hours | Oral, IV | Preferred in asthmatics |
| Atenolol | Cardioselective (β1) | 6-9 hours | Oral | Renal excretion |
| Esmolol | Cardioselective (β1) | 9 MINUTES | IV only | Ultra-short acting, used in acute settings, perioperatively |
| Sotalol | Non-selective + K+ block | 12 hours | Oral | Also Class III! |
| Carvedilol | β1 + β2 + α1 block | 7-10 hours | Oral | Used in heart failure |
Esmolol is particularly important to remember: its 9-minute half-life (metabolized by red blood cell esterases) makes it ideal for acute rate control - you can stop it quickly if needed.
Adverse Effects
- Bradycardia - too slow heart rate
- Heart block - can worsen pre-existing AV conduction disease
- Bronchospasm - especially non-selective agents (block β2 in bronchial smooth muscle). CONTRAINDICATED in asthma and COPD (use cardioselective agents with caution)
- Heart failure precipitation - negative inotropic effect (avoid in decompensated heart failure; but chronic use in stable HF is beneficial)
- Cold extremities, Raynaud's - peripheral vasoconstriction
- Masked hypoglycemia in diabetics - symptoms of hypoglycemia (tachycardia, tremor) are blocked; sweating is NOT masked (important distinction)
- Fatigue, sleep disturbances, depression (especially with lipophilic agents like propranolol)
- Impotence, sexual dysfunction
- Lipid abnormalities - increased triglycerides, decreased HDL
Contraindications
- Severe bradycardia or heart block (without pacemaker)
- Decompensated heart failure
- Asthma (non-selective agents absolutely contraindicated)
- Cocaine toxicity (non-selective beta-blockers contraindicated - can cause unopposed alpha stimulation and severe hypertension/coronary vasospasm)
- Pheochromocytoma (unless combined with alpha-blockade first)
CLASS III ANTIARRHYTHMICS
K+ Channel Blockers: Amiodarone, Sotalol, Dofetilide, Ibutilide, Dronedarone
Definition
Class III antiarrhythmics block K+ channels (predominantly Ikr - the rapidly activating delayed rectifier K+ current), prolonging Phase 3 repolarization and thus extending the action potential duration (APD) and effective refractory period (ERP).
Mechanism of Action
K+ channel blockade → K+ efflux slows → Phase 3 takes longer → APD and ERP prolonged → more time before the next impulse can come → anti-re-entry effect
ECG effect: Prolonged QT interval (QRS width is unchanged, but the ST segment and T wave are longer)
The prolonged ERP means:
- In re-entry: the circling impulse arrives and finds tissue still refractory → circuit terminates
- Rate of spontaneous firing decreases
The major risk: Prolonging Phase 3 too much → during the long repolarization, small inward currents can trigger an Early Afterdepolarization (EAD) → triggers Torsades de Pointes
AMIODARONE - "The Most Effective and Most Toxic Antiarrhythmic"
Amiodarone deserves its own extended section. It is unique because it has ALL FOUR classes of antiarrhythmic activity:
| Action | Class | Clinical Significance |
|---|
| Na+ channel block | Class I | Slows conduction |
| Beta-blockade | Class II | Slows SA and AV node |
| K+ channel block | Class III | Prolongs APD, ERP - main antiarrhythmic effect |
| Ca2+ channel block | Class IV | Further slows SA, AV node |
Additionally, amiodarone structurally resembles thyroid hormone and interferes with thyroid metabolism significantly.
Clinical Uses (extremely broad):
- AF and atrial flutter - rate AND rhythm control
- Ventricular tachycardia (VT) and ventricular fibrillation (VF) - the drug of choice for sustained VT/VF in patients with structural heart disease
- Pulseless VT/VF in cardiac arrest - IV amiodarone (ACLS protocol)
- Post-cardiac surgery arrhythmia prevention
- AF in heart failure patients (amiodarone and dofetilide are the only antiarrhythmics shown not to increase mortality in heart failure)
Pharmacokinetics - Highly Unusual:
- Extremely lipophilic - concentrates massively in fat, liver, lungs, skin, thyroid
- Half-life: 40-55 DAYS (range 15-142 days) - the longest half-life of any drug in common use
- Loading dose required (oral: typically 200-400 mg 3x/day for 1-2 weeks, then maintenance 100-400 mg/day)
- Takes weeks to reach steady state; takes weeks to be eliminated after stopping
- Low bioavailability (~40%) - variable absorption
- Inhibits CYP2D6, CYP3A4, and P-glycoprotein → many drug interactions
Adverse Effects - Comprehensive (EXTREMELY HIGH YIELD):
The multiple toxicities of amiodarone are a favorite exam topic. Every organ system can be affected.
| System | Adverse Effect | Details |
|---|
| Pulmonary | Pulmonary toxicity (pneumonitis/fibrosis) | 1-15% of patients. Dry cough, dyspnea, pulmonary infiltrates on CXR, decreased DLCO. Can be fatal. Monitor CXR and PFTs every 12 months. |
| Thyroid | Both hypo- AND hyperthyroidism | 2-5% per year. Amiodarone contains 37% iodine by weight. Hypothyroidism (more common in iodine-sufficient areas). Hyperthyroidism (more common in iodine-deficient areas). Monitor TSH every 6 months. |
| Eyes | Corneal microdeposits | Virtually 100% of patients on long-term therapy. Usually asymptomatic. Visible on slit lamp. Not a reason to stop drug unless causing visual symptoms. Rare optic neuritis can cause blindness. |
| Skin | Photosensitivity + blue-gray discoloration | Common. Blue-gray skin (especially sun-exposed areas) may not resolve on stopping drug. |
| Liver | Hepatotoxicity | Transaminase elevation common. Monitor ALT/AST every 6 months. Stop if >3x normal. |
| Neurological | Peripheral neuropathy, ataxia, tremor | Relatively common with long-term use |
| Cardiac | Bradycardia, heart block, QT prolongation | TdP is actually RARE despite QT prolongation (amiodarone prolongs APD uniformly, reducing TdP risk) |
| Thyroid | Note: amiodarone can INTERFERE with thyroid tests even without true dysfunction | |
The BLUE-GRAY skin and corneal deposits are among the most photographed exam questions. If you see a patient with chronic arrhythmia and blue-gray skin, think AMIODARONE.
Drug Interactions (critical):
- Warfarin: Amiodarone inhibits CYP2C9 → increases warfarin levels dramatically → increase bleeding risk → reduce warfarin dose by 30-50% when starting amiodarone
- Digoxin: Amiodarone increases digoxin levels by reducing its renal excretion and displacing it from tissue proteins → reduce digoxin dose by 50%
- Many other interactions via CYP inhibition
Why does amiodarone cause less TdP than other Class III drugs despite QT prolongation?
- Other Class III drugs cause heterogeneous QT prolongation (some areas of the heart repolarize later than others, creating dispersion of repolarization - the substrate for TdP)
- Amiodarone causes homogeneous prolongation of repolarization throughout the heart, so dispersion is minimal
- This is called "amiodarone paradox" - prolonged QT but low TdP risk
Monitoring Protocol for Amiodarone (high-yield for viva/clinicals):
- Baseline: ECG, TFT (TSH), LFTs, CXR, PFTs, eye examination
- Every 6 months: TFT, LFTs, clinical assessment for pulmonary symptoms
- Every 12 months: CXR, PFTs
- Eye exam if any visual symptoms
SOTALOL
Mechanism: Non-selective beta-blocker (Class II) + K+ channel block (Class III)
This dual mechanism makes sotalol unique: it slows the heart rate (like a beta-blocker) AND prolongs the refractory period (like Class III).
Clinical Uses:
- AF and atrial flutter
- Life-threatening ventricular tachycardia/fibrillation
- Maintenance of sinus rhythm after cardioversion of AF
Adverse Effects:
- Beta-blocker effects: Bradycardia, bronchospasm, fatigue (CONTRAINDICATED in asthma)
- Class III effects: QT prolongation → TdP (more prone than amiodarone because it causes heterogeneous prolongation)
- TdP risk is GREATER at SLOWER heart rates (reverse-use-dependent block - the channels are blocked more when the heart beats slowly, which is the opposite of use-dependent and is actually dangerous)
Important: Sotalol requires QTc monitoring. If QTc > 500 ms on treatment, stop the drug.
DOFETILIDE
Mechanism: Pure K+ channel blocker (specifically Ikr) - the most selective Class III drug
Clinical Uses:
- AF and atrial flutter, especially in patients with heart failure (one of only two antiarrhythmics not shown to increase mortality in HF - the other being amiodarone)
- Cardioversion of AF to sinus rhythm
Key Points:
- Renally excreted - dose must be reduced in renal failure
- Due to high TdP risk, dofetilide initiation requires in-hospital monitoring for at least 3 days with continuous QTc monitoring and dose adjustment
- Patients are NOT discharged until dofetilide dose is stabilized and QTc is safe
IBUTILIDE
Mechanism: K+ channel block (Class III) + activates slow inward Na+ current → prolongs action potential
Clinical Uses:
- IV only
- Acute cardioversion of recent-onset AF and atrial flutter to sinus rhythm
- Most effective for flutter (conversion rate ~60-70%)
Critical adverse effect: HIGH risk of TdP (up to 4% of patients). After IV ibutilide, patients MUST be monitored for at least 4 hours.
DRONEDARONE
Mechanism: Similar to amiodarone - has Class I, II, III, IV properties. But it does NOT contain iodine (amiodarone's iodine content is responsible for thyroid and some other toxicities).
Advantages over amiodarone: No thyroid toxicity, no pulmonary toxicity, no corneal deposits, shorter half-life (24 hours)
Disadvantages: Less effective than amiodarone; liver toxicity including severe hepatic failure requiring transplant has been reported
Critical contraindication: CONTRAINDICATED in permanent AF with heart failure - the PALLAS trial showed increased mortality when dronedarone was used in permanent AF with cardiovascular risk factors.
Clinical uses: Non-permanent AF in patients with normal LV function, as an alternative to amiodarone for maintaining sinus rhythm
Class III Summary Table
| Drug | Mechanism | Route | Special Toxicity | Use |
|---|
| Amiodarone | Class I+II+III+IV | Oral, IV | Multi-organ (thyroid, lung, liver, skin, cornea) | Broadest - VT/VF, AF |
| Sotalol | Beta-block + K+ block | Oral | TdP, bronchospasm | AF, VT |
| Dofetilide | Pure K+ block | Oral | High TdP risk, renal dosing | AF (HF patients) |
| Ibutilide | K+ block + Na+ | IV only | Very high TdP (4%) | Acute AF/flutter cardioversion |
| Dronedarone | I+II+III+IV (no iodine) | Oral | Hepatotoxicity; ↑ mortality in permanent AF with HF | Non-permanent AF |
CLASS IV ANTIARRHYTHMICS
Non-Dihydropyridine Ca2+ Channel Blockers: Verapamil and Diltiazem
Definition
Class IV antiarrhythmics block L-type (long-lasting) voltage-gated Ca2+ channels in the heart, predominantly affecting the SA node and AV node.
Why ONLY the SA and AV Nodes?
Remember: the SA and AV nodes are "slow response" cells - they rely on Ca2+ channels (not Na+ channels) for their action potential upstroke.
Ventricular and atrial cells use fast Na+ channels predominantly.
So blocking Ca2+ channels has the most impact on SA and AV nodes.
Mechanism of Action
L-type Ca2+ channel block in SA and AV nodes:
- SA node: Slows spontaneous Phase 4 depolarization AND slows Phase 0 upstroke → slower heart rate (negative chronotropy)
- AV node: Slows Phase 0 of AV nodal cells → slower AV conduction → prolonged PR interval → fewer impulses get through to ventricles (rate control) → negative dromotropic effect
- Myocardium: Reduced Ca2+ entry → reduced contractility (negative inotropic effect)
ECG effects: Bradycardia + prolonged PR interval (same as beta-blockers but mechanism is different)
Class IV Drugs: Verapamil vs. Diltiazem
| Feature | Verapamil | Diltiazem |
|---|
| Type | Phenylalkylamine | Benzothiazepine |
| Heart effect | More negative inotrope | Less negative inotrope |
| Vasodilation | Less | More |
| Half-life | 6-8 hours | 3-4 hours (varies with formulation) |
| Routes | Oral, IV | Oral, IV |
| Use | SVT, rate control AF, HOCM | SVT, rate control AF, angina |
| Special toxicity | Constipation, gingival hyperplasia | Less constipation |
Remember: Dihydropyridine Ca2+ channel blockers (nifedipine, amlodipine) are used for hypertension and angina. They act mainly on peripheral blood vessels. They have much less cardiac effect and are NOT antiarrhythmics.
Clinical Uses
- Rate control in AF and atrial flutter (reduce ventricular rate)
- Termination of AVNRT (paroxysmal SVT) - IV verapamil or diltiazem can terminate most SVTs within minutes by blocking the AV nodal component of the re-entry circuit
- Stable angina (diltiazem)
- HOCM (verapamil reduces outflow obstruction)
Adverse Effects
- Bradycardia and AV block - most important cardiac effect
- Negative inotropic effect - avoid in systolic heart failure (verapamil especially dangerous)
- Hypotension
- Constipation - verapamil is famous for this; sometimes used to treat diarrhea
- Peripheral edema (especially with diltiazem)
- Gingival hyperplasia - verapamil (like cyclosporine and nifedipine)
- Gynecomastia - verapamil (rarely)
Critical Drug Interaction: Verapamil + Beta-Blockers = DANGEROUS
Verapamil and beta-blockers both slow the SA and AV nodes. Combining them can cause:
- Severe bradycardia
- Complete AV block
- Asystole (cardiac standstill)
They should never be given together IV (can be used together orally with caution, but monitoring required).
Another critical interaction: Verapamil + Digoxin → verapamil increases digoxin levels → digoxin toxicity
Critical Contraindication: Verapamil is CONTRAINDICATED in WPW + AF
This is a classic exam trap:
In WPW syndrome, there is an accessory pathway (e.g., Bundle of Kent) between atria and ventricles. When AF occurs in WPW:
- Normally, the AV node acts as a "gatekeeper" - it limits how fast impulses reach the ventricles
- Verapamil/diltiazem slow the AV node further, BUT they can paradoxically SPEED UP conduction through the accessory pathway
- This can lead to extremely rapid ventricular rates (300+ bpm) and ventricular fibrillation
In WPW with AF: Use procainamide or cardioversion. AVOID verapamil, diltiazem, beta-blockers, and digoxin.
UNCLASSIFIED ANTIARRHYTHMICS
ADENOSINE
Mechanism:
- Adenosine binds to A1 receptors in the AV node
- This activates a specific K+ channel (IKAch - acetylcholine-sensitive K+ channel)
- K+ efflux hyperpolarizes the AV nodal cell → makes it harder to excite
- Effectively causes a transient, complete AV block for about 10-30 seconds
- Also inhibits cAMP generation → reduces Ca2+ entry
Clinical Uses:
- DRUG OF CHOICE for acute termination of paroxysmal SVT (PSVT/AVNRT) - by transiently blocking the AV node, it interrupts the re-entry circuit and terminates the SVT
- Diagnostic tool - helps distinguish SVT from VT (VT is not terminated by adenosine; SVT usually is)
- Not effective for AF or flutter (they don't depend on AV nodal conduction for the arrhythmia itself)
Administration: Rapid IV bolus (6 mg first dose, then 12 mg if no response) followed immediately by rapid saline flush. Must be injected fast because adenosine has an extremely short half-life (< 10 seconds - metabolized by red blood cells and vascular endothelium). Must be given through a large, proximal vein.
Adverse Effects:
- Flushing - very common, harmless
- Chest tightness and dyspnea - brief bronchospasm; AVOID in asthma (use verapamil instead)
- Transient sense of doom - patients often describe a brief, terrifying sensation of impending death (caused by the brief AV block)
- Transient AV block, sinus pause - these are expected and brief
Key Facts:
- Theophylline (and caffeine) are adenosine ANTAGONISTS - they block A1 receptors. A patient who just had coffee may need a higher dose of adenosine.
- Dipyridamole (blocks adenosine reuptake) POTENTIATES adenosine - a smaller dose is needed.
- Adenosine is safe in WPW (it only blocks AV node, not the accessory pathway) for diagnostic purposes.
DIGOXIN (Cardiac Glycoside)
Mechanism:
- Primary mechanism: Inhibits Na+/K+-ATPase (the sodium pump)
- Na+ accumulates inside the cell → Na+/Ca2+ exchanger works less → Ca2+ inside the cell increases
- Higher intracellular Ca2+ → increased contractility (positive inotropic effect)
- Indirect vagotonic effect (stimulates the vagus nerve) → slows SA node and AV node
Clinical Uses:
- AF: Rate control (slows ventricular rate by increasing AV nodal block via vagus nerve)
- Heart failure with reduced EF (positive inotropic effect - increases contractility; improves symptoms and reduces hospitalization, but does NOT reduce mortality)
- Atrial flutter (rate control)
The Narrow Therapeutic Index - Critical:
- Therapeutic range: 0.5-2.0 ng/mL
- Toxic level: >2.0-2.5 ng/mL
- Very easy to overdose
Factors that increase digoxin toxicity risk:
- Hypokalemia (MOST IMPORTANT): K+ and digoxin compete for the Na+/K+-ATPase binding site. If K+ is low, more digoxin binds → enhanced effect/toxicity. This is why diuretic-induced hypokalemia is dangerous in patients on digoxin.
- Hypomagnesemia
- Hypercalcemia
- Hypothyroidism
- Renal failure (digoxin is renally excreted; dose must be reduced)
- Old age (reduced renal clearance)
- Drug interactions: Amiodarone, quinidine, verapamil all increase digoxin levels
Digoxin Toxicity - Signs and Symptoms:
- GI: Anorexia, nausea, vomiting (earliest and most common)
- CNS: Confusion, delirium, visual disturbances (classic: yellow-green vision - xanthopsia, or halos around lights - chromatopsia)
- Cardiac: Almost ANY arrhythmia! But most characteristic: paroxysmal atrial tachycardia with AV block (PAT with block), ventricular bigeminy, AV block
- In children: Sinus bradycardia is often the first sign
Treatment of Digoxin Toxicity:
- Stop digoxin
- Correct hypokalemia and hypomagnesemia
- For bradycardia/AV block: Atropine or pacing
- For ventricular arrhythmias: Lidocaine or phenytoin (NOT quinidine or Class IC - they can worsen)
- For severe toxicity: Digoxin-specific antibody fragments (Digibind/DigiFab) - the antidote
MAGNESIUM SULFATE
Mechanism: Mechanism not fully elucidated; may stabilize membranes, affect ion channels
Clinical Uses:
- Treatment of Torsades de Pointes - IV magnesium is the first-line treatment for TdP, even if serum Mg2+ is normal
- Digoxin-induced arrhythmias
- Hypomagnesemia-associated arrhythmias
SECTION 4: TEACH USING ANALOGIES
Analogies to Make This Stick Forever
The Orchestra Analogy
The heart is a perfectly coordinated orchestra. The SA node is the conductor. Every musician (cardiac cell) watches the conductor and plays in sequence. An arrhythmia is what happens when:
- A random musician starts playing out of turn (abnormal automaticity)
- A sound bounces between the walls and creates an echo that keeps repeating (re-entry)
- A musician gets startled by a loud sound and plays an extra note (triggered activity)
Antiarrhythmic drugs are like the rules the concert hall enforces to prevent chaos.
Class I Drugs: The Door Analogy
Na+ channels are like swinging doors that let sodium rush into the cell. Class I drugs put a LOCK on the door - varying speed and strength:
- Class IA: A padlock - moderately slows entry, AND makes the hallway longer (prolongs Phase 3). Think "moderate lock that also lengthens the corridor."
- Class IB: A weak magnet - barely locks, AND actually shortens the hallway. Works best on "broken" (ischemic) doors.
- Class IC: A DEADBOLT - slam! Door barely opens at all. Great in healthy corridors, CATASTROPHIC in damaged ones.
Class III Drugs: The Hotel Room Analogy
After a cardiac cell fires, it must "recover" (repolarize) before it can fire again - like a hotel room that must be cleaned before the next guest.
Class III drugs slow down the cleaning crew (block K+ channels) → the room takes longer to be ready → the next impulse has to WAIT → re-entry circuits break because they arrive to find the room still being cleaned.
Amiodarone is like a thorough cleaning company that also fixes pipes, replaces lightbulbs, checks the HVAC, and repaints - it does everything (Class I+II+III+IV) but it also damages the building over time (multi-organ toxicity).
The Reentry Circuit: Broken Ring Road
Imagine a ring road around a city. Normally, cars enter, go around, and exit. A unidirectional road block means cars can only go clockwise. Normally this would mean cars accumulate. But if a back road exists, some cars loop backwards on it - and now they keep driving in circles forever. Antiarrhythmic drugs either:
- Block the back road more completely (Class I - bidirectional block)
- Put up a "road closed for maintenance" sign for longer (Class III - prolong refractory period so cars arrive and find the road closed)
Amiodarone: The Good Chemotherapy
Like chemotherapy, amiodarone is the most effective option for the worst arrhythmias, but it comes at a price - multi-organ toxicity with long-term use. You use it when you need it most, monitor carefully, and use the minimum effective dose.
Lidocaine: The Firefighter Who Only Works Where There's Fire
Lidocaine preferentially acts on ischemic (depolarized) tissue - exactly where the fire (arrhythmia source) is. It barely touches healthy tissue. This targeted action is why it is so useful for post-MI arrhythmias.
Digoxin: The Strong-Willed Helper With a Temper
Digoxin strengthens the heart (positive inotropic effect) and slows the rate, but it has a "temper" - the therapeutic index is so narrow that it can easily cause the very arrhythmias it is meant to prevent. And low potassium makes it even more volatile.
SECTION 5: STEP-BY-STEP CLINICAL REASONING
Case 1: Patient with Atrial Fibrillation
Patient: 65-year-old man with hypertension and palpitations. ECG shows irregularly irregular rhythm, no P waves - confirmed AF.
Step 1: What are the GOALS of treatment in AF?
- Rate control (slow the ventricular rate to < 110 bpm at rest)
- Rhythm control (restore and maintain sinus rhythm)
- Anticoagulation (prevent stroke)
Step 2: Rate control - Which drug?
- For most patients: Beta-blocker (metoprolol) or non-dihydropyridine Ca2+ channel blocker (diltiazem, verapamil)
- In heart failure with reduced EF: Beta-blocker or digoxin (Ca2+ channel blockers with negative inotropy are dangerous here)
- In COPD/asthma: Diltiazem or verapamil (not beta-blockers)
- Rapid effect needed? IV metoprolol or IV diltiazem
Step 3: Rhythm control - Which drug?
- No structural heart disease: Flecainide or propafenone (Class IC) - well-tolerated "pill-in-pocket" option
- Structural heart disease (HF, LVH, CAD): Amiodarone (safe, effective) OR dofetilide (in HF)
- Post-MI (structural heart disease): NEVER use Class IC (CAST trial!)
Step 4: Can we cardiovert?
- AF < 48 hours: Can cardiovert without prolonged anticoagulation
- AF > 48 hours: Anticoagulate for 3 weeks before cardioversion (or do TEE to rule out atrial thrombus)
- Pre-cardioversion antiarrhythmic loading to maintain sinus rhythm: Amiodarone, propafenone, or flecainide
Summary of AF drug choice:
AF Patient
├── Rate control:
│ ├── Normal LV function → β-blocker or Ca²⁺ blocker
│ └── HF → β-blocker or digoxin
└── Rhythm control:
├── No structural disease → Flecainide/Propafenone/Sotalol/Dronedarone
└── Structural disease (HF, CAD, LVH) → AMIODARONE or Dofetilide
Case 2: Narrow Complex Tachycardia (SVT/AVNRT) in Emergency
Patient: 30-year-old woman with sudden palpitations, HR 190 bpm, narrow QRS on ECG.
Step 1: Try vagal maneuvers first
- Valsalva maneuver (bear down), carotid sinus massage
- Increases vagal tone → slows AV node → may terminate SVT
Step 2: If vagal maneuvers fail → Adenosine
- Give 6 mg rapid IV bolus (followed by fast saline flush) into a large antecubital vein
- This blocks AV node for 10-30 seconds
- In AVNRT (most common SVT): This terminates the re-entry circuit through the AV node
- Success rate ~90%
Step 3: If adenosine fails
- Give 12 mg adenosine (repeat once)
- Or switch to IV verapamil or IV diltiazem
Step 4: If patient is hemodynamically unstable (hypotension, chest pain, collapse)
- SKIP medications
- Immediate synchronized DC cardioversion
Step 5: Prevention of recurrent SVT
- Catheter ablation (definitive cure for AVNRT)
- Long-term: Verapamil, diltiazem, or beta-blockers
Case 3: Post-MI Patient With Frequent PVCs
Patient: 55-year-old man, 3 days post-MI, multiple PVCs on telemetry.
Step 1: Are these PVCs dangerous?
- PVCs post-MI can degenerate into VT/VF - they represent the substrate for re-entry in ischemic tissue
Step 2: Drug choice?
- Beta-blocker (metoprolol) - FIRST LINE. Reduces mortality post-MI, prevents VT/VF
- NOT Class IC drugs (CAST trial - increases mortality)
- NOT quinidine (no mortality benefit, proarrhythmic)
- For established VT: IV lidocaine or amiodarone
Step 3: If the patient develops sustained VT or VF?
- Unstable → IMMEDIATE defibrillation
- Pulseless VT/VF → CPR + defibrillation + IV amiodarone (300 mg) or IV lidocaine
Case 4: Torsades de Pointes
Patient: Woman on QT-prolonging antibiotics (azithromycin) + sotalol. QTc 540 ms. Develops polymorphic VT that self-terminates repeatedly.
Step 1: Recognize TdP
- QT prolongation + polymorphic VT with characteristic "twisting of points" pattern on ECG
Step 2: Immediate treatment
- IV MAGNESIUM SULFATE (2 g bolus) - first-line even if Mg2+ is normal
- Remove the offending drug (stop sotalol, stop azithromycin)
- Correct electrolytes (correct hypokalemia, hypomagnesemia)
Step 3: If TdP persists
- Increase heart rate: Atropine (IV) or temporary pacing at 90-110 bpm (faster rate shortens QT interval and prevents TdP)
- Isoproterenol infusion (increases rate, shortens QT)
Step 4: Prevention
- Avoid QT-prolonging drugs
- Correct electrolytes before using antiarrhythmics
- Genetic testing for congenital long QT syndrome
SECTION 6: MEMORY TOOLS
Mnemonic 1: Class IA Drugs - "Queen PD Has Anticholinergic Effects"
Quinidine, Procainamide, Disopyramide
- Quinidine: Cinchonism, Digoxin interaction (↑levels)
- Procainamide: Lupus (drug-induced), Slow acetylators more prone
- Disopyramide: Urinary retention (anticholinergic), Heart failure risk
Mnemonic 2: Procainamide = SLENA
- Slow acetylators more prone to lupus
- Lupus-like syndrome (SLE) - unique toxicity
- Extended active metabolite: NAPA (Class III)
- NAPA is the active metabolite
- Agranulocytosis (rare but serious)
Mnemonic 3: Class IB "Ischemic heart's BFF" - Lidocaine and Mexiletine
- Works best on ischemic (depolarized, inactivated) tissue
- Brain toxicity for lidocaine (NOT heart)
- Fast kinetics
- First choice post-MI acute VT
Mnemonic 4: Class IC - "CAST ME OUT of ischemic hearts"
- CAST trial showed they INCREASE mortality in structural heart disease
- Marked QRS widening
- Exclude from any structural heart disease patient
- Use only in pristine (structurally normal) hearts for AF
Mnemonic 5: Amiodarone Side Effects - "THYROID PLATE"
- Thyroid (hypo AND hyperthyroidism)
- Hepatotoxicity
- Yellow/Blue: corneal deposits + blue-gray skin
- Respiratory: pulmonary toxicity/fibrosis
- Ophthalmologic: optic neuritis, corneal deposits
- Interacts: warfarin (↑), digoxin (↑)
- Depolarization: slows SA, AV nodes
- Photosensitivity
- Long half-life (40-55 days)
- All four Vaughan-Williams class actions
- The best antiarrhythmic (most effective)
- Every organ can be affected
Mnemonic 6: Class III K+ blockers - "SAD DI"
- Sotalol (also beta-blocker)
- Amiodarone
- Dofetilide
- Dronedarone
- Ibutilide (IV only, acute AF)
Mnemonic 7: Drugs That Prolong QT (Risk of TdP) - "MACHO QT"
- Macrolide antibiotics (azithromycin)
- Antiarrhythmics Class IA and III
- Cisapride (prokinetic - withdrawn in many countries)
- Haloperidol and antipsychotics
- Other: fluoroquinolones, tricyclics, antihistamines (terfenadine - withdrawn)
- Quinidine specifically (Class IA)
- Triggered by hypokalemia, hypomagnesemia, bradycardia
Mnemonic 8: Digoxin Toxicity Signs - "NAVY GI"
- Nausea/Vomiting (GI - first sign)
- Arrhythmias (any type; PAT with block is classic)
- Vision changes (yellow-green xanthopsia, halos)
- Yellow-green vision (xanthopsia)
- GI upset
- Inhibited Na/K-ATPase by digoxin
Drug Comparison Table: The Big Picture
| Drug Class | Na+ | K+ | Ca2+ | β-block | APD | QRS | QT | Best Use | Worst Danger |
|---|
| IA (Quinidine) | ++ | ++ | - | - | ↑ | Wide | ↑ | AF/VT | TdP, cinchonism |
| IA (Procainamide) | ++ | + | - | - | ↑ | Wide | ↑ | WPW+AF, VT | Drug-induced lupus |
| IA (Disopyramide) | ++ | ++ | - | - | ↑ | Wide | ↑ | AF, HOCM | Urinary retention, HF |
| IB (Lidocaine) | ++ | - | - | - | ↓ | Min | ↓ | Acute VT/VF post-MI | CNS toxicity (seizures) |
| IB (Mexiletine) | ++ | - | - | - | ↓ | Min | ↓ | Chronic VT | Nausea/vomiting |
| IC (Flecainide) | +++ | - | - | - | ~= | Wide↑↑ | ~= | AF (no struct. disease) | Proarrhythmia, ↑ mortality in MI |
| II (Metoprolol) | - | - | - | ++ | ~= | ~= | ~= | Post-MI, AF rate, SVT | Bronchospasm, bradycardia |
| III (Amiodarone) | ++ | +++ | ++ | ++ | ↑↑ | ~= | ↑ | VT/VF, AF, HF+AF | Multi-organ toxicity |
| III (Sotalol) | - | +++ | - | ++ | ↑ | ~= | ↑ | AF, VT | TdP, bronchospasm |
| III (Dofetilide) | - | +++ | - | - | ↑ | ~= | ↑ | AF in HF | TdP, renal dosing |
| IV (Verapamil) | - | - | ++ | - | ~= | ~= | ~= | SVT, AF rate | HF, bradycardia, constipation |
| Adenosine | - | - | - | - | ~= | ~= | ~= | Acute SVT | Flushing, bronchospasm |
| Digoxin | - | - | - | ~= | - | ~= | ~= | AF rate, HF | Narrow TI, hypokalemia TDI |
Quick Visual: Where Each Class Acts on the Action Potential
+30 mV ─────────────────╮
Phase 1\ Phase 2 (Plateau)
─────────────────────╮
0 mV \
\─── Phase 3
-90 mV ────────────────────────────────────────────────────────
Phase 4 (flat in ventricle / slopes in SA node)
CLASS IA = Slows Phase 0 upstroke + Prolongs Phase 3
CLASS IB = Slightly slows Phase 0 + Shortens Phase 3
CLASS IC = Markedly slows Phase 0 (minimal Phase 3 effect)
CLASS II = Decreases Phase 4 slope (SA/AV node)
CLASS III = Prolongs Phase 3 (K+ channel block)
CLASS IV = Slows Phase 0 of SA/AV node (Ca2+ dependent)
SECTION 7: EXAMINER'S CORNER
Most Likely Essay Questions (MBBS Final Exams)
-
"Classify antiarrhythmic drugs. Describe the mechanism of action, clinical uses, and adverse effects of amiodarone."
- Answer structure: Vaughan-Williams classification (table) → amiodarone: Class I+II+III+IV mechanism → uses (VT, VF, AF, AF in HF) → toxicities (thyroid, pulmonary, hepatic, skin, corneal, neurological) → monitoring
-
"Describe the pharmacological basis and management of atrial fibrillation."
- Answer structure: Background on AF (re-entry, AV node) → rate control drugs (beta-blockers, Ca2+ blockers, digoxin) → rhythm control (flecainide/propafenone in structurally normal heart; amiodarone in structural disease) → CAST trial → anticoagulation
-
"Write short notes on: (a) Drug-induced lupus, (b) Torsades de Pointes, (c) Lidocaine toxicity."
-
"Describe the mechanism of action and clinical uses of adenosine as an antiarrhythmic drug."
-
"What are the mechanisms by which cardiac arrhythmias arise? How do antiarrhythmic drugs act to suppress them?"
Most Likely Short Notes
- Amiodarone - mechanism and toxicity
- Lidocaine as antiarrhythmic
- Drug-induced lupus (procainamide)
- Digoxin toxicity and treatment
- Torsades de Pointes
- CAST trial and its significance
- Adenosine in SVT
- Class III antiarrhythmics
- QT prolongation by drugs
- Verapamil vs. diltiazem
Most Likely Viva Questions
-
"What is the mechanism of action of lidocaine? Why is it not given orally?"
- Na+ channel block; IV only because of massive first-pass metabolism in liver; first-pass hepatic extraction ~70%
-
"Why is flecainide contraindicated after myocardial infarction?"
- CAST trial showed increased mortality; in ischemic tissue, Class IC drugs create NEW re-entry circuits and cause proarrhythmia
-
"What is the most serious adverse effect of procainamide? Who is more prone?"
- Drug-induced lupus (DILE); slow acetylators are more prone
-
"How does amiodarone cause thyroid toxicity?"
- Contains 37% iodine by weight; chronic iodine load; also structurally resembles T4 and inhibits thyroid hormone binding/conversion. Both hypo- and hyperthyroidism possible.
-
"What is the antidote for digoxin toxicity?"
- Digoxin-specific antibody fragments (Fab fragments) - Digibind/DigiFab
-
"Why is verapamil contraindicated in WPW with AF?"
- WPW has accessory pathway bypassing AV node. Verapamil blocks AV node but can accelerate conduction through the accessory pathway → very rapid ventricular rate → VF.
-
"What is the mechanism of quinidine syncope?"
- QT prolongation → Torsades de Pointes → loss of consciousness (syncope)
-
"Which drug is used for Torsades de Pointes?"
- IV Magnesium Sulfate (2g bolus) is first-line
-
"What are the ECG effects of Class I antiarrhythmics?"
- Class IA: Widened QRS + prolonged QT
- Class IB: Minimal ECG change (may shorten QT)
- Class IC: Markedly widened QRS
-
"Why does amiodarone rarely cause TdP despite prolonging the QT interval?"
- Amiodarone prolongs APD homogeneously (uniformly) throughout the myocardium → minimal dispersion of repolarization → low TdP risk. Other Class III drugs cause heterogeneous prolongation → high TdP risk.
Most Likely MCQs with Traps
MCQ 1: A patient on digoxin is started on quinidine. What adjustment must be made?
- A) Increase digoxin dose
- B) Reduce digoxin dose by half ← CORRECT
- C) Stop digoxin
- D) No change needed
- Trap: Students forget that quinidine both displaces digoxin from tissue proteins AND reduces its renal clearance, doubling blood levels.
MCQ 2: Which antiarrhythmic should be AVOIDED in asthma?
- A) Verapamil
- B) Adenosine
- C) Sotalol ← CORRECT (also D is true but between them sotalol is most specifically contraindicated as antiarrhythmic)
- D) Propafenone
- Explanation: Sotalol has both beta-blocking and K+ channel blocking actions; its beta-blockade causes bronchospasm. Adenosine also causes bronchospasm; propafenone also has weak beta-blocking effects. In a strict MCQ context, sotalol and propafenone are specifically contra-indicated in asthma. Verapamil is preferred for SVT in asthmatic patients.
MCQ 3: A 55-year-old post-MI patient has frequent PVCs. Which drug is CONTRAINDICATED?
- A) Metoprolol
- B) Amiodarone
- C) Flecainide ← CORRECT
- D) Lidocaine
- Trap: CAST trial - Class IC drugs (flecainide, encainide) increase mortality in post-MI structural heart disease.
MCQ 4: What is the first sign of lidocaine toxicity?
- A) Nystagmus ← CORRECT
- B) Seizures
- C) Cardiac arrest
- D) Hypotension
- Trap: Students jump to seizures (the most dramatic toxicity), but nystagmus is the EARLIEST sign.
MCQ 5: Which drug causes a lupus-like syndrome as an adverse effect?
- A) Quinidine
- B) Disopyramide
- C) Procainamide ← CORRECT
- D) Lidocaine
MCQ 6: Adenosine's mechanism in terminating SVT involves:
- A) Na+ channel blockade
- B) Activation of K+ channels via A1 receptors (IKAch) ← CORRECT
- C) Beta-adrenergic blockade
- D) Ca2+ channel blockade
MCQ 7: Quinidine's ability to cause QT prolongation is due to blockade of:
- A) Na+ channels
- B) K+ channels (Ikr) ← CORRECT
- C) Ca2+ channels
- D) Beta receptors
MCQ 8: Disopyramide is useful in which of the following conditions (besides arrhythmias)?
- A) Heart failure
- B) BPH
- C) Hypertrophic Obstructive Cardiomyopathy (HOCM) ← CORRECT
- D) Asthma
- Trap: Disopyramide is negative inotropic which is HARMFUL in heart failure but USEFUL in HOCM (reduces outflow obstruction).
MCQ 9: The drug of choice for terminating Torsades de Pointes is:
- A) Lidocaine
- B) IV Magnesium Sulfate ← CORRECT
- C) Amiodarone
- D) Quinidine
- Trap: Students choose amiodarone (the "powerful" antiarrhythmic), but TdP requires magnesium first. Note: DO NOT use Class IA or Class III drugs (they prolong QT further!).
MCQ 10: Which of the following antiarrhythmics is safe to use in patients with atrial fibrillation AND heart failure with reduced ejection fraction?
- A) Flecainide
- B) Dronedarone
- C) Amiodarone ← CORRECT (also dofetilide, but amiodarone is more commonly tested)
- D) Propafenone
Common Student Traps in Examinations
-
Saying amiodarone causes TdP frequently - It PROLONGS QT but RARELY causes TdP (due to homogeneous prolongation)
-
Forgetting that sotalol has BOTH Class II AND Class III properties - It is a beta-blocker with K+ channel blocking activity
-
Saying lidocaine is effective for atrial arrhythmias - It is NOT; it only works for ventricular arrhythmias
-
Forgetting the CAST trial implications - If a question involves post-MI patient with arrhythmias, Class IC is wrong
-
Treating WPW + AF with verapamil - DANGEROUS. This is a classic killer question.
-
Confusing procainamide and quinidine drug interactions with digoxin - BOTH increase digoxin levels. Quinidine AND amiodarone both increase digoxin. Learn all three.
-
Saying procainamide lupus is the same as SLE - Drug-induced lupus is ANA-positive, anti-histone antibody positive, but DOES NOT cause nephritis or CNS disease (unlike true SLE). It REVERSES on stopping the drug.
-
Forgetting that mexiletine is the oral form of lidocaine's class (IB) - and that GI side effects are its main toxicity (not CNS)
-
Using Class IA or Class III drugs to treat Torsades de Pointes - These prolong QT and will WORSEN TdP. Magnesium is the answer.
-
Forgetting that dronedarone is contraindicated in permanent AF with HF - The PALLAS trial showed increased mortality in this population.
SECTION 8: SPECIAL TOPICS
The CAST Trial - Why It Changed Everything (1989)
Before 1989, the thinking was: "Ventricular arrhythmias after MI → suppress them with antiarrhythmics → prevent sudden death."
The CAST trial tested flecainide and encainide in post-MI patients with asymptomatic ventricular ectopy.
Result: Sudden death was DOUBLED in the flecainide/encainide group compared to placebo.
Lesson: Suppressing a surrogate marker (ectopic beats) does not always improve outcomes. Antiarrhythmic drugs can be proarrhythmic. The drug that silences a rhythm problem on paper can create a deadlier one in the ischemic substrate.
This trial established that not all arrhythmias need treatment and that structural heart disease is a contraindication to Class IC drugs.
Drug-Induced QT Prolongation and Torsades de Pointes
This is an extremely important drug safety issue in clinical practice:
Common drugs that prolong QT (beyond antiarrhythmics):
- Macrolide antibiotics: Azithromycin, erythromycin
- Fluoroquinolones: Ciprofloxacin, moxifloxacin
- Antifungals: Fluconazole
- Antipsychotics: Haloperidol, chlorpromazine, ziprasidone
- Tricyclic antidepressants
- Antiemetics: Metoclopramide, droperidol
- Antihistamines: Terfenadine, astemizole (withdrawn due to TdP risk)
Risk factors for drug-induced TdP:
- Baseline QT prolongation
- Female sex (women have slightly longer QT at baseline)
- Hypokalemia and hypomagnesemia
- Bradycardia (slower rate = longer QT)
- Structural heart disease
- High drug concentrations (overdose, renal failure, CYP inhibitor co-administration)
Prevention:
- Check baseline QTc before starting
- Correct electrolytes
- Avoid combinations of multiple QT-prolonging drugs
- Monitor ECG regularly
WPW Syndrome and Antiarrhythmics
WPW (Wolff-Parkinson-White) syndrome involves an accessory pathway (Bundle of Kent) that bypasses the AV node. This has critical implications for drug choice:
In WPW with SVT (without AF):
- Adenosine or verapamil may be used
- The re-entry circuit can go: AV node down → bundle of Kent back up (orthodromic)
In WPW with AF (pre-excited AF):
- The accessory pathway conducts extremely rapidly (no decremental conduction like the AV node)
- Drugs that BLOCK the AV node (verapamil, diltiazem, beta-blockers, digoxin, adenosine) will drive ALL impulses down the accessory pathway → extremely rapid ventricular rate → VF → death
The safe drugs in WPW+AF: Procainamide (IV), cardioversion
The dangerous drugs in WPW+AF: Verapamil, diltiazem, beta-blockers, digoxin, adenosine
SECTION 9: HIGH-YIELD REVISION SHEET
One-Page Rapid Review: Antiarrhythmics
Must-Know Classification
CLASS IA: Quinidine, Procainamide, Disopyramide
→ Na+ block (moderate) + K+ block → Wide QRS + Long QT
→ Quinidine: cinchonism, ↑digoxin levels
→ Procainamide: DRUG-INDUCED LUPUS (slow acetylators), NAPA metabolite
→ Disopyramide: urinary retention (anticholinergic), HF risk, useful in HOCM
CLASS IB: Lidocaine (IV), Mexiletine (oral)
→ Na+ block (fast kinetics) → preferential ischemic tissue → shorten APD
→ Lidocaine: CNS toxicity (nystagmus → seizures), IV only
→ Mexiletine: GI toxicity (nausea/vomiting)
→ ONLY for ventricular arrhythmias; NOT atrial
CLASS IC: Flecainide, Propafenone
→ Na+ block (slow, potent) → markedly wide QRS
→ CONTRAINDICATED in structural heart disease (CAST trial - ↑mortality in MI)
→ Propafenone: also weak beta-block → bronchospasm
→ Safe only in structurally NORMAL hearts (AF/flutter)
CLASS II: Beta-blockers (Metoprolol, Atenolol, Esmolol, Propranolol)
→ Slow Phase 4 (SA node) → bradycardia
→ Prolong PR (AV node block)
→ POST-MI: drug of choice; SVT; rate control in AF
→ Esmolol: half-life 9 min (IV, perioperative)
CLASS III: K+ channel blockers
→ Prolong Phase 3 → long QT → anti-re-entry
→ Amiodarone: BEST antiarrhythmic; Class I+II+III+IV
Toxicity: THYROID, PULMONARY, LIVER, SKIN (blue-gray + photosensitivity), CORNEAL DEPOSITS
Half-life: 40-55 DAYS; ↑warfarin; ↑digoxin
→ Sotalol: beta-block + K+ block; TdP risk; AVOID in asthma
→ Dofetilide: pure K+ block; safe in HF; high TdP; renal dosing
→ Ibutilide: IV; acute AF/flutter cardioversion; HIGH TdP risk (4%)
→ Dronedarone: no iodine, no thyroid/lung toxicity; CONTRAINDICATED in permanent AF + HF
CLASS IV: Verapamil, Diltiazem (non-DHP CCBs)
→ Block Ca2+ channels in SA/AV nodes
→ Rate control in AF; terminate SVT
→ CONTRAINDICATED in WPW+AF, systolic HF (verapamil especially), + beta-blockers IV
→ Verapamil: constipation, gingival hyperplasia
Must-Know Toxicities
| Drug | Unique Toxicity |
|---|
| Quinidine | Cinchonism, increases digoxin levels |
| Procainamide | Drug-induced lupus (SLE), anti-histone Abs |
| Disopyramide | Anticholinergic (urinary retention, dry mouth) |
| Lidocaine | CNS toxicity: nystagmus, seizures (NOT cardiac) |
| Flecainide/IC | Proarrhythmia; increased mortality post-MI (CAST) |
| Amiodarone | Thyroid + pulmonary + liver + corneal + blue-gray skin |
| Sotalol | TdP, bronchospasm |
| Ibutilide | TdP (4% incidence) |
| Digoxin | Yellow vision, PAT with block, toxic on hypokalemia |
| Adenosine | Flushing, chest tightness, transient AV block, bronchospasm |
Must-Know Drug Interactions
| Offending Drug | Object Drug | Effect | Action |
|---|
| Quinidine | Digoxin | ↑ digoxin 2x | Halve digoxin dose |
| Amiodarone | Warfarin | ↑ anticoagulation (↑ bleeding) | Reduce warfarin ~30-50% |
| Amiodarone | Digoxin | ↑ digoxin levels | Halve digoxin dose |
| Verapamil | Digoxin | ↑ digoxin levels | Monitor closely |
| Verapamil | Beta-blockers (IV) | Severe bradycardia, asystole | AVOID combination IV |
| CYP2D6 inhibitors | Mexiletine | ↑ mexiletine toxicity | Caution |
Exam Emergency Facts
- Adenosine → Drug of choice for acute SVT (rapid IV bolus, short half-life < 10 sec)
- Magnesium sulfate → Drug of choice for Torsades de Pointes
- Procainamide → Drug-induced lupus (slow acetylators more prone)
- Lidocaine → IV only; CNS toxicity (seizures); ONLY for ventricular arrhythmias
- Amiodarone → Most effective; all 4 classes; multi-organ toxicity; half-life 40-55 days
- Flecainide/Class IC → CONTRAINDICATED post-MI (CAST trial)
- Verapamil → CONTRAINDICATED in WPW+AF (causes VF) and in systolic HF
- Digoxin → Toxic in hypokalemia; antidote = Digibind (Fab fragments)
- Sotalol → Class II + III; AVOID in asthma (beta-blockade)
- Post-MI arrhythmia prevention → Beta-blockers (NOT Class IC)
SECTION 10: SELF-ASSESSMENT
10 Short-Answer Questions with Explanations
Question 1: A 40-year-old woman presents with palpitations. ECG shows narrow complex tachycardia at 185 bpm. Vagal maneuvers fail. What is the drug of choice for acute termination, and what is the mechanism?
Answer: Adenosine (6 mg rapid IV bolus, followed by 12 mg if needed).
Mechanism: Adenosine binds A1 receptors on AV nodal cells, activating the acetylcholine-sensitive K+ channel (IKAch). This hyperpolarizes the AV nodal cell, causing transient complete AV block (~10-30 seconds). Since AVNRT (the most common SVT) requires AV nodal conduction for its re-entry circuit, blocking the AV node terminates the circuit. Half-life < 10 seconds means effects are brief and self-limiting.
Question 2: A patient is being started on amiodarone for ventricular tachycardia. He is also taking warfarin (INR 2.5). What monitoring is required and why?
Answer: Amiodarone inhibits CYP2C9, which is responsible for metabolizing warfarin. This raises warfarin blood levels significantly, increasing the anticoagulant effect and bleeding risk. The INR will rise above therapeutic range. Warfarin dose should be empirically reduced by 30-50% when amiodarone is started, and INR should be monitored closely every 1-2 weeks until stable. Additionally, since amiodarone has a half-life of 40-55 days, its enzyme-inhibitory effect persists for weeks to months even after stopping amiodarone - warfarin dose adjustment must be maintained during this period.
Question 3: Why is lidocaine NOT given orally as an antiarrhythmic drug?
Answer: Lidocaine undergoes extensive first-pass metabolism in the liver (hepatic extraction ratio approximately 70%). When given orally, most of the drug is metabolized before it reaches the systemic circulation, resulting in negligible plasma levels and no antiarrhythmic effect. Oral administration is therefore therapeutically useless. For oral therapy of ventricular arrhythmias in the same class, mexiletine (a structurally similar Class IB drug) is used instead, as it is resistant to first-pass metabolism.
Question 4: A 50-year-old man with a previous myocardial infarction has atrial fibrillation. Why is flecainide contraindicated? What drugs would you use instead?
Answer: Flecainide (Class IC) is contraindicated because post-MI patients have structural heart disease with ischemic scar tissue. The CAST trial (1989) demonstrated that flecainide and other Class IC drugs increased the rate of sudden cardiac death in post-MI patients with ventricular ectopy. The mechanism is proarrhythmia: marked Na+ channel blockade slows conduction in ischemic tissue so severely that new re-entry circuits are created, leading to fatal ventricular arrhythmias.
Safer alternatives: For AF rate control in post-MI: beta-blockers or digoxin. For rhythm control (sinus rhythm maintenance): Amiodarone is the drug of choice in structural heart disease. Dofetilide is an alternative, particularly in heart failure.
Question 5: What is Torsades de Pointes? How do you treat it?
Answer: Torsades de Pointes (TdP) is a form of polymorphic ventricular tachycardia (VT) associated with a prolonged QT interval. On ECG, the QRS complexes appear to "twist" around the isoelectric baseline - the amplitude gradually increases then decreases over 5-20 beats, giving the characteristic morphology. It often self-terminates but can degenerate into ventricular fibrillation.
Mechanism: QT prolongation (from drugs, electrolyte disturbances, or congenital long QT syndrome) creates a long vulnerable period during repolarization. Early afterdepolarizations (EADs) occur during the prolonged Phase 2-3 and can trigger a new action potential, initiating TdP.
Treatment:
- IV Magnesium Sulfate (2 g bolus) - first-line, even if Mg2+ is normal. Stabilizes the myocardium and suppresses EADs.
- Remove causative drug (stop offending antiarrhythmic, antibiotic, etc.)
- Correct electrolytes (especially K+; keep K+ > 4.5 mEq/L)
- If persistent: Temporary pacing or isoproterenol to increase heart rate (faster rate shortens QT interval, preventing TdP)
- If unstable: Immediate defibrillation
DO NOT use: Class IA or Class III drugs (they further prolong QT).
Question 6: What is the unique adverse effect of procainamide? What antibody is associated with it?
Answer: Procainamide causes drug-induced lupus erythematosus (DILE). With chronic use, 25-30% of patients develop this syndrome. Features include arthralgia, arthritis, pleuritis, pericarditis, and skin rash - mimicking systemic lupus erythematosus.
Antibodies: Nearly all patients on long-term procainamide develop positive ANA (antinuclear antibodies). The antibody most specifically associated is anti-histone antibody - this is characteristic of drug-induced lupus and helps distinguish it from idiopathic SLE.
Important distinctions from true SLE:
- Drug-induced lupus does NOT typically cause glomerulonephritis (renal disease)
- Drug-induced lupus does NOT cause CNS manifestations
- It is reversible on stopping the drug
- Anti-dsDNA antibodies (characteristic of true SLE) are usually NEGATIVE in drug-induced lupus
Who is most at risk? Slow acetylators - they metabolize procainamide more slowly, resulting in higher drug accumulation and greater lupus risk.
Question 7: A patient with WPW syndrome develops atrial fibrillation. Which drugs are DANGEROUS? Which is safe?
Answer: WPW syndrome involves an accessory pathway (Bundle of Kent) that bypasses the AV node. In AF, multiple atrial impulses (400-600/min) are generated. Normally, the AV node acts as a gatekeeper, allowing only 100-180/min through to the ventricles.
DANGEROUS drugs:
- Verapamil and Diltiazem - block AV node but can paradoxically enhance conduction through the accessory pathway, allowing extremely rapid ventricular rates (300+ bpm) → VF → death
- Beta-blockers - same principle (block AV node, may increase accessory pathway conduction)
- Digoxin - shortens accessory pathway refractory period, accelerating conduction through it
- Adenosine - similar concern (though it is sometimes used diagnostically with caution)
Safe and preferred treatment:
- IV Procainamide - blocks the accessory pathway conduction AND AV node conduction (drug of choice for WPW+AF in hemodynamically stable patient)
- Synchronized DC cardioversion - if hemodynamically unstable
Question 8: Compare the adverse effects of verapamil and diltiazem.
Answer:
| Feature | Verapamil | Diltiazem |
|---|
| Negative inotropy | MORE prominent | Less |
| Constipation | PROMINENT (characteristic) | Less common |
| Gingival hyperplasia | Present (rare) | Not significant |
| AV block | Both can cause | Both can cause |
| Hypotension | Both | Both |
| Peripheral edema | Less | More prominent |
| Gynecomastia | Reported (rare) | Not characteristic |
| Heart failure risk | Higher (avoid in systolic HF) | Slightly lower risk |
Common to both: Bradycardia, AV block, hypotension, dizziness. Both are contraindicated in WPW+AF, decompensated systolic heart failure, severe bradycardia/heart block, and should not be combined with IV beta-blockers.
Question 9: A patient develops visual changes and yellow-green colored vision. They have bradycardia and ECG shows paroxysmal atrial tachycardia with AV block. What drug is responsible and how do you treat?
Answer: This is classic digoxin toxicity.
Diagnosis:
- Xanthopsia (yellow-green vision) - the famous visual side effect, described by some painters (notably Van Gogh is theorized to have been on digitalis)
- Nausea/vomiting - usually the first symptoms
- PAT (paroxysmal atrial tachycardia) with AV block - the most pathognomonic ECG finding of digoxin toxicity (enhanced automaticity of atrial tissue + increased AV block)
- Bradycardia - vagotonic effect + Na+/K+-ATPase inhibition in SA node
Treatment:
- STOP digoxin immediately
- Correct hypokalemia (most common precipitant) - give IV KCl carefully
- Correct hypomagnesemia
- For bradycardia/AV block: Atropine (0.5 mg IV); temporary pacing if unresponsive
- For ventricular arrhythmias: Lidocaine or phenytoin (NOT quinidine - it increases digoxin levels further!)
- For severe/life-threatening toxicity: Digoxin-specific Fab antibody fragments (Digibind/DigiFab) - the specific antidote. Each vial binds ~0.5 mg digoxin. Dose calculated based on ingested amount or serum digoxin level.
Question 10: What is the half-life of amiodarone? Why is it clinically significant?
Answer: The half-life of amiodarone is 40-55 days (range 15-142 days). This is the longest half-life of any commonly used drug, and it has several major clinical implications:
-
Loading dose required: Because the drug distributes so extensively into fat, liver, lungs, and other tissues (enormous volume of distribution ~60 L/kg), it takes weeks to reach effective plasma and tissue levels. A loading regimen is used to rapidly achieve therapeutic levels.
-
Slow onset of action: Even with a loading dose, full antiarrhythmic effect may take days to weeks.
-
Drug interactions persist long after stopping: Amiodarone's inhibition of CYP2C9 (affecting warfarin) and CYP3A4 continues for months after the drug is discontinued. This means the drug may continue to raise warfarin levels for months after stopping.
-
Adverse effects persist: If a patient develops pulmonary toxicity or thyroid dysfunction, the drug continues to accumulate (and remains in tissues) for months even after stopping. Toxicity may worsen briefly after discontinuation.
-
Cannot quickly adjust: Unlike drugs with short half-lives (e.g., esmolol: 9 minutes), amiodarone cannot be titrated rapidly. Once started, it takes months to fully wash out.
-
Monitoring must continue months after stopping: Because the drug persists, organ monitoring (thyroid, liver, lungs) must continue well beyond drug discontinuation.
This comprehensive learning note was compiled from: Lippincott Illustrated Reviews: Pharmacology; Goodman & Gilman's The Pharmacological Basis of Therapeutics; Harrison's Principles of Internal Medicine 22e (2025); Katzung's Basic and Clinical Pharmacology 16e; Braunwald's Heart Disease; Goldman-Cecil Medicine; The Washington Manual of Medical Therapeutics.
End of Antiarrhythmics Complete Learning Note