Haemolytic anaemia in detail its types and pathology Strictly acc to robbins pathology
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Finding Sources
Loading Image
Finding Sources
I have all the content I need. Now I'll compose the comprehensive answer strictly from Robbins & Kumar Basic Pathology (and Robbins, Cotran & Kumar Pathologic Basis of Disease where relevant).
Haemolytic Anaemia - Strictly According to Robbins Pathology
Robbins & Kumar Basic Pathology (Robbins Pathology), 10th ed., ISBN 9780323790185
Definition and Overview
Haemolytic anaemias are a diverse group of disorders sharing a common feature - accelerated red cell destruction. The red cell lifespan is shortened to less than its normal 120 days, often markedly so. The resulting anaemia and low tissue O₂ stimulate erythropoietin release, leading to:
- Marrow erythroid hyperplasia
- Peripheral blood reticulocytosis (hallmarks of haemolytic anaemia)
- In severe cases: extramedullary haematopoiesis in liver, spleen, and lymph nodes
Classification
Robbins provides two principal ways to classify haemolytic anaemias:
1. By Origin of Defect
| Category | Description | Examples |
|---|---|---|
| Intrinsic (intracorpuscular) | Defect within the red cell itself | Hereditary spherocytosis, Sickle cell, Thalassemia, G6PD deficiency, PNH |
| Extrinsic (extracorpuscular) | External factor causes destruction | Immunohemolytic anaemia, Mechanical haemolysis, Malaria |
2. By Site of Haemolysis (Clinically more useful)
Extravascular Haemolysis
Caused by defects that increase destruction of red cells by phagocytes, particularly in the spleen. The spleen sequesters non-deformable or antibody-coated cells; macrophages in splenic cords phagocytose them.
Distinctive findings:
- Hyperbilirubinaemia and jaundice (from Hb degradation in macrophages)
- Splenomegaly (work hyperplasia of phagocytes)
- Cholelithiasis with pigment (bilirubin-rich) gallstones if long-standing
- Low serum haptoglobin (macrophages regurgitate enough Hb to deplete it)
- No iron deficiency (iron recycling by phagocytes is efficient)
Intravascular Haemolysis
Red cells burst within the circulation due to mechanical forces, complement fixation, or biochemical damage (clostridial toxins, heat).
Distinctive findings:
- Haemoglobinaemia - free Hb in plasma
- Haemoglobinuria - Hb passes into urine (Hb molecule is small enough)
- Haemosiderinuria - iron accumulation in renal tubular cells shed into urine
- Iron deficiency - iron is lost via urine rather than recycled
- Low serum haptoglobin (also seen in extravascular)
Types of Haemolytic Anaemia
1. Hereditary Spherocytosis
Transmission: Autosomal dominant (most common); rare severe autosomal recessive form.
Pathogenesis: Inherited defects in the membrane skeleton proteins that stabilize the lipid bilayer.
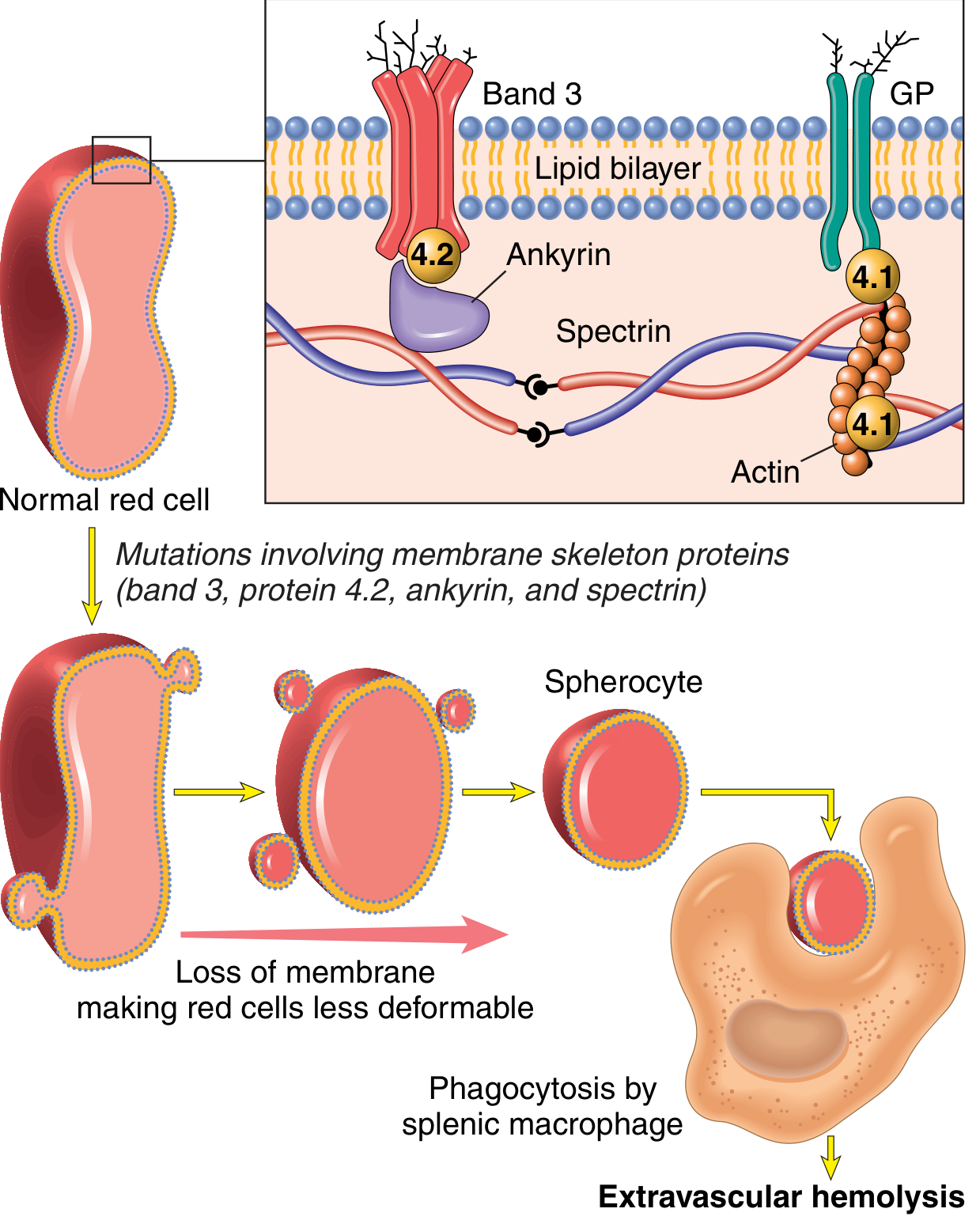
The membrane skeleton is a meshwork of:
- Spectrin (major protein) - long, flexible heterodimer
- Actin filaments
- Ankyrin and band 4.1 - linker proteins
- Connected to intrinsic membrane proteins band 3 and glycophorin
Mutations weaken interactions between the membrane skeleton and intrinsic membrane proteins → lipid bilayer destabilizes → red cells shed membrane vesicles → surface area-to-volume ratio decreases → spherocytes form.
Mechanism of haemolysis: Spherocytes have limited deformability → sequestered in splenic cords → destroyed by resident macrophages (extravascular haemolysis). Splenectomy corrects the anaemia despite persistence of spherocytes.
Morphology:
- Peripheral smear: spherocytes are dark red and lack central pallor
- Compensatory marrow erythroid hyperplasia + reticulocytosis
- Splenomegaly (most prominent of all haemolytic anaemias) - splenic weight 500-1000 g (normal 150-200 g)
- Marked congestion of splenic cords, increased macrophages
- Cholelithiasis in 40-50% of patients
Treatment: Splenectomy corrects the anaemia; must weigh against infection risk from encapsulated bacteria (especially in children).
2. Sickle Cell Anaemia
The prototypic haemoglobinopathy. Most common familial haemolytic anaemia.
Genetics: Single amino acid substitution in β-globin - valine replaces glutamate at the 6th position. HbS allele is prevalent where falciparum malaria was endemic (equatorial Africa, India, southern Europe, Middle East). In the USA, ~8% of African-descent individuals are HbS carriers; ~1 in 600 have sickle cell anaemia.
Pathogenesis:
- HbS differs from HbA: valine instead of glutamate at β-globin position 6
- On deoxygenation, HbS molecules undergo conformational change → polymers form via intermolecular contacts involving the abnormal valine → red cells assume elongated crescentic (sickle) shape
- Sickling is initially reversible on reoxygenation; with repeated episodes → membrane damage (calcium influx, K⁺ and water loss) → irreversibly sickled cells → haemolysis
Three factors governing sickling:
- Intracellular levels of non-HbS Hb - HbA retards HbS polymerization greatly → HbS heterozygotes (sickle cell trait) have little sickling in vivo. HbF also retards polymerization → newborns asymptomatic until HbF falls (~5-6 months). HbC (lysine instead of glutamate) interacts with HbS only moderately → compound heterozygotes have milder disease
- Intracellular Hb concentration - high MCHC favours polymerization; dehydration worsens sickling
- Length of time in deoxygenated state - slow blood flow prolongs deoxygenation
Morphology:
- Peripheral smear: sickle-shaped red cells, target cells, nucleated red cells
- Vaso-occlusive crises - hallmark; microinfarcts in bones, spleen, liver, brain, lungs, penis
- Spleen: initially enlarged by red pulp congestion with sickled cells; progressive splenic infarction leads to autosplenectomy (fibrotic, shrunken spleen by adulthood)
- Severe splenomegaly can occur in compound heterozygotes (HbSC)
- Bone marrow hyperplasia → cortical bone thinning, "crew cut" appearance on skull X-ray
- Increased risk of aplastic crisis (Parvovirus B19), sequestration crisis, infections by encapsulated bacteria (functional asplenia)
Treatment: Hydroxyurea reduces crises by:
- Increasing HbF levels
- Anti-inflammatory effect (inhibits WBC production)
- Increases red cell size, lowering intracellular Hb concentration
- Metabolizes to NO (vasodilator, inhibits platelet aggregation)
3. Thalassemia
Thalassemias are inherited disorders caused by mutations in globin genes that decrease the synthesis of α- or β-globin. Deficiency of Hb + intracellular precipitates from excess unpaired normal chain → red cell damage and haemolysis. Prevalent in Mediterranean, African, and Asian regions (protection against falciparum malaria).
Genetics: Autosomal codominant. α-globin: 2 genes on chromosome 16. β-globin: single gene on chromosome 11.
β-Thalassemia
| Type | Genotype | Clinical Features |
|---|---|---|
| β-Thalassemia major | Homozygous | Severe anaemia; requires regular transfusions |
| β-Thalassemia intermedia | Variable | Moderately severe; transfusions not always required |
| β-Thalassemia minor | Heterozygous | Mild microcytic anaemia; usually asymptomatic |
Pathogenesis:
- Point mutations impair transcription, splicing, or translation of β-globin mRNA
- Excess α-globin chains form insoluble precipitates → damage red cell membranes → ineffective erythropoiesis (destruction of erythroid precursors in marrow) + haemolysis
- Both extravascular haemolysis and ineffective erythropoiesis contribute to anaemia
Morphology (β-Thalassemia major):
- Marked microcytosis, hypochromia, poikilocytosis, anisocytosis, nucleated red cells (normoblasts)
- Target cells (increased surface area-to-volume ratio)
- Striking erythroid hyperplasia filling intramedullary space → cortical bone thinning, impaired growth, skeletal deformities
- Extramedullary haematopoiesis → prominent splenomegaly, hepatomegaly, lymphadenopathy
- Growth retardation and cachexia
- Severe haemosiderosis from iron overload (transfusions + increased gut iron absorption due to suppressed hepcidin from expanded erythropoiesis)
Clinical features:
- β-Thalassemia minor/trait: asymptomatic, normal life expectancy; mild microcytic hypochromic anaemia
- β-Thalassemia major: manifests postnatally as HbF diminishes; growth retardation from infancy; survival into 2nd-3rd decade with transfusions, but iron overload develops; chelation therapy (deferoxamine) + bone marrow transplant can be curative
α-Thalassemia
- Caused by deletion of α-globin genes (usually entire genes deleted)
- Severity depends on number of genes deleted (0-4):
- 1 gene deleted: silent carrier state
- 2 genes deleted: α-thalassemia trait - mild microcytic anaemia
- 3 genes deleted: HbH disease - excess β-chains form β₄ tetramers (HbH); relatively stable but high O₂ affinity (poor O₂ delivery)
- 4 genes deleted: Hydrops fetalis - lethal in utero; excess γ-chains form γ₄ (Hb Bart); near-zero O₂ delivery capacity
4. Glucose-6-Phosphate Dehydrogenase (G6PD) Deficiency
Gene: X chromosome (X-linked). >400 G6PD variants identified; few associated with disease.
Pathogenesis: Red cells are constantly exposed to oxidants, normally inactivated by reduced glutathione (GSH). G6PD is essential for GSH synthesis. In G6PD deficiency:
- Oxidant stress (from drugs, infections, foods) overwhelms deficient GSH
- Oxidants attack globin chains → oxidized haemoglobin denatures → precipitates as Heinz bodies (intracellular inclusions)
- Heinz bodies damage the red cell membrane → intravascular haemolysis
- Cells with less damage lose deformability; splenic macrophages "pluck out" Heinz bodies → bite cells (Fig. 10.6) → trapped and destroyed in spleen
Trigger agents:
- Antimalarials (primaquine), sulfonamides, nitrofurantoin, phenacetin, high-dose aspirin, vitamin K derivatives
- Fava beans (favism)
- Infections (most common trigger - phagocytes generate oxidants as host response)
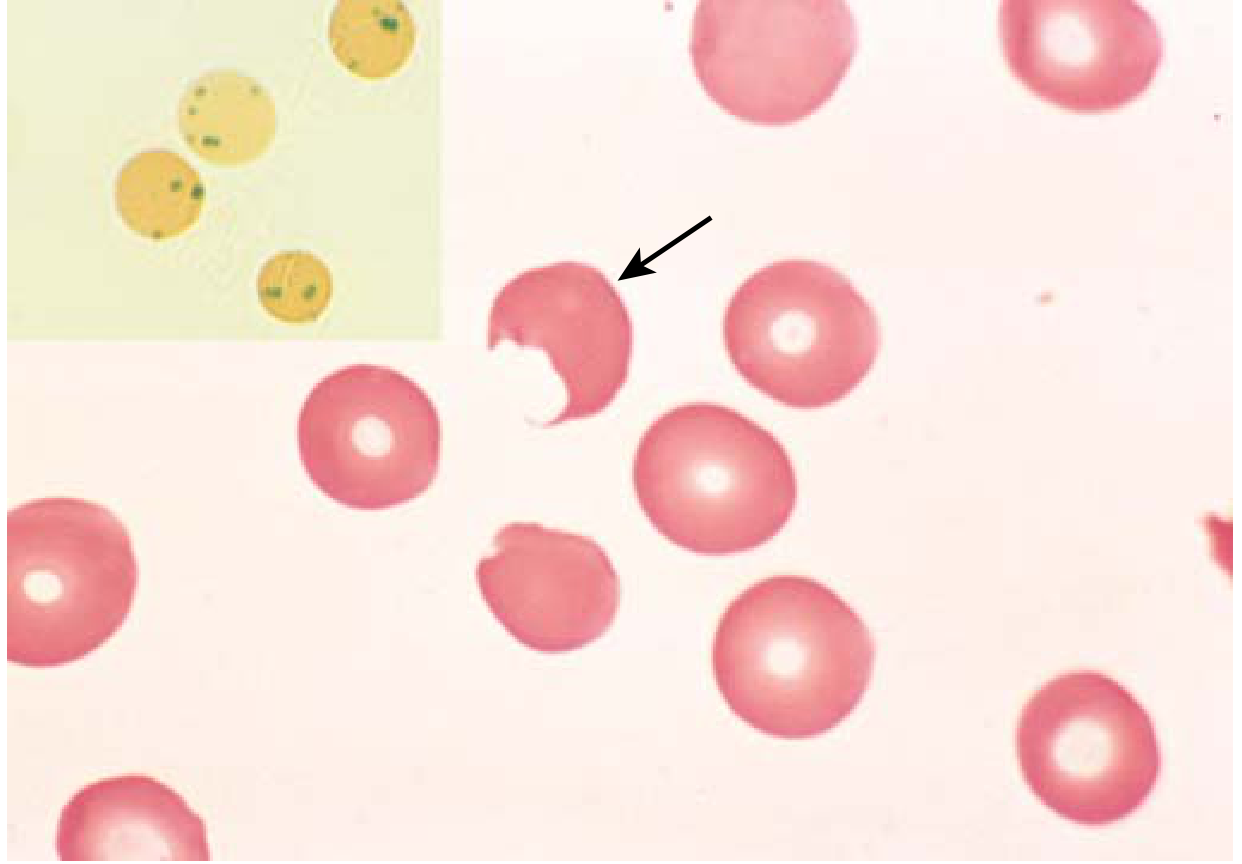
Clinical features:
- Haemolysis 2-3 days after drug exposure; variable severity
- Males uniformly affected (X-linked)
- Heterozygous females: two RBC populations due to lyonization; most unaffected unless "unfavorable lyonization" (large proportion of deficient cells)
- G6PD A- variant (Africa): only older red cells lysed (modest enzyme decrease); haemolysis self-limited as marrow replaces with new cells with adequate G6PD
- G6PD Mediterranean (Middle East): more marked deficiency; more severe haemolysis
5. Paroxysmal Nocturnal Haemoglobinuria (PNH)
Pathogenesis: Acquired mutation in PIG-A gene (encodes an enzyme required for synthesis of GPI anchors) in a haematopoietic stem cell → clonal expansion → red cells, WBCs, and platelets lacking GPI-anchored proteins, including CD55 (decay-accelerating factor) and CD59 (protectin). These proteins normally inhibit complement on self-cell surfaces. Their absence → unregulated complement activation → intravascular haemolysis via membrane attack complex (C5b-C9).
Clinical features:
- Classic (but uncommon) presentation: nocturnal haemolysis (sleep-related CO₂ retention → decreased pH → enhanced complement fixation)
- Most present with chronic anaemia and iron deficiency from chronic intravascular haemolysis
- Association with aplastic anaemia (may precede or follow PNH)
- Most feared complication: thrombosis in abdominal vessels (portal vein, hepatic vein) - related to excessive complement activity
Treatment - Eculizumab: Anti-C5 antibody that inhibits MAC assembly → lessens intravascular haemolysis and thrombosis dramatically. However:
- Does NOT affect early complement fixation → C3b continues to deposit → continuing extravascular haemolysis
- Blocks C5b-C9 → risk of Neisseria (meningococcal) infections → all patients must be vaccinated against N. meningococcus
6. Immunohemolytic Anaemia
Caused by antibodies binding to antigens on red cell membranes. May arise spontaneously or be drug-induced.
Diagnosis: Direct Coombs test - patient's red cells + anti-human Ig/complement antibodies → agglutination indicates Ig/complement coating.
Warm Antibody Type
- IgG (rarely IgA) antibodies active at 37°C
-
60% idiopathic; 25% in immunologic disorders (SLE) or drug-induced
- Mechanism: IgG-coated cells phagocytosed in spleen; also "nibbling" by macrophages removes membrane → spherocytes → rapid splenic destruction (same as hereditary spherocytosis)
- Drug mechanisms:
- α-methyldopa: induces autoantibodies against Rh blood group antigens
- Penicillin: binds covalently to red cell membrane proteins → neoantigens → antibody response
- Some drugs form immune complexes that deposit on red cells → fix complement or act as opsonins
Cold Antibody Type
Classification of Immunohemolytic Anaemias (Table 10.4):
| Warm Antibody Type | Cold Antibody Type |
|---|---|
| Primary (idiopathic) | Acute: Mycoplasma pneumonia, infectious mononucleosis |
| Secondary: B-cell neoplasms (CLL), autoimmune (SLE), drugs (α-methyldopa, penicillin, quinidine) | Chronic: idiopathic, B-cell lymphoid neoplasms (lymphoplasmacytic lymphoma) |
- Low-affinity IgM antibodies binding at temperatures <30°C (e.g., distal extremities in cold)
- Transient forms: Mycoplasma pneumonia, infectious mononucleosis (mild, clinically unimportant)
- Chronic forms: B-cell neoplasms or idiopathic
- Pathogenesis: IgM initiates complement fixation; later steps inefficient at <37°C → cells coated with C3b and C3d but not lysed. In warm areas, IgM released but C3b/C3d remain → phagocytosis by macrophages (mainly spleen and liver) → mostly extravascular haemolysis
- IgM pentavalency → crosslinks red cells → agglutination → sludging in capillaries → Raynaud phenomenon
7. Mechanical Haemolysis
Two major settings:
-
Traumatic (macroangiopathic) haemolysis:
- Dysfunctional cardiac valve prostheses - turbulence creates "blender effect"
- Repeated physical pounding (marathon racing, karate, bongo drumming)
-
Microangiopathic haemolytic anaemia (MAHA):
- Small vessels partially obstructed by fibrin strands, platelet thrombi, or other lesions → passing red cells are sheared into schistocytes (helmet cells, triangular cells)
- Causes: DIC (most common - fibrin deposition), severe hypertension, thrombotic thrombocytopenic purpura (TTP), haemolytic-uraemic syndrome (HUS)
8. Malaria
- Haemolysis caused by Plasmodium parasites invading and destroying red cells
- P. falciparum is the most severe form; protective pressure explains why HbS, G6PD deficiency, and thalassemia mutations are prevalent in malaria-endemic areas
- Haemolysis is both intravascular (rupture of parasitized cells) and extravascular (phagocytosis of parasitized cells)
Summary Table
| Type | Mechanism | Intrinsic/Extrinsic | Site of Haemolysis | Key Morphology |
|---|---|---|---|---|
| Hereditary Spherocytosis | Membrane skeleton defect (spectrin, ankyrin, band 3) | Intrinsic | Extravascular (spleen) | Spherocytes, no central pallor |
| Sickle Cell | HbS polymerization on deoxygenation | Intrinsic | Extra + intravascular | Sickle cells, target cells |
| β-Thalassemia major | Excess α-chain precipitation, ineffective erythropoiesis | Intrinsic | Mainly extravascular | Microcytes, hypochromia, poikilocytosis, nucleated RBCs |
| G6PD Deficiency | Oxidant-induced Heinz body formation | Intrinsic | Extra + intravascular | Heinz bodies, bite cells |
| PNH | Absence of CD55/CD59, complement MAC | Intrinsic (acquired) | Intravascular | No characteristic RBC morphology |
| Warm Antibody IHA | IgG opsonization, spherocyte formation | Extrinsic | Extravascular (spleen) | Spherocytes, + Coombs |
| Cold Antibody IHA | IgM + C3b opsonization | Extrinsic | Mainly extravascular | Agglutination, Raynaud phenomenon |
| MAHA | Physical shearing by fibrin/platelet thrombi | Extrinsic | Intravascular | Schistocytes (helmet cells) |
| Malaria | Parasite invasion/rupture | Extrinsic | Extra + intravascular | Parasitized RBCs |
All content strictly from: Robbins & Kumar Basic Pathology (Robbins Pathology), 10th ed. (ISBN 9780323790185), Chapter 10 - Red Cell Disorders, pp. 347-356.
This is a shared conversation. Sign in to Orris to start your own chat.