Explain the pathophysiology of tuberculosis
Reading File
Finding Sources
Finding Sources
Searching PubMed
"tuberculosis"[MeSH Terms] AND "pathogenesis"[MeSH Terms]
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Searching Images
tuberculosis granuloma pathophysiology
Reading File
I now have comprehensive material to write a thorough answer. Let me compile it with the textbook figures.
Pathophysiology of Tuberculosis
1. The Causative Organism
Mycobacterium tuberculosis (MTB) is a slow-growing, obligate aerobe and obligate intracellular pathogen. Its defining structural feature is a uniquely thick, lipid-rich cell wall containing mycolic acids, lipoarabinomannan (LAM), and sulfolipids. This wall confers:
- Acid-fastness (resistance to decolorization by acid-alcohol)
- Resistance to phagocytic digestion
- Promotion of cough through sulfolipid-mediated irritation, enhancing transmissibility
MTB is classified into distinct phylogenetic lineages. "Modern" lineages 2–4 drive the global epidemic; certain lineage 2 (Beijing) strains are more virulent, more transmissible, and more prone to acquiring drug resistance.
2. Transmission and Entry
Infection begins with inhalation of aerosolized droplet nuclei (1–5 μm) expelled by an individual with active pulmonary TB. These particles are small enough to bypass mucociliary defenses and deposit directly in the distal alveoli. Cavitary disease (harboring 10⁷–10⁹ bacilli) is the most infectious form. On average, one untreated infectious individual infects 3–10 people per year.
3. Innate Immune Response and Macrophage Evasion
Upon reaching alveoli, MTB is phagocytosed by alveolar macrophages — the first line of defense. However, MTB has evolved multiple strategies to subvert killing:
- Phagosome maturation arrest: MTB inhibits fusion of the phagosome with lysosomes, preventing acidification and exposure to degradative enzymes. It does this via lipoarabinomannan (LAM), which blocks Ca²⁺/calmodulin signaling required for phagolysosome fusion.
- Antioxidant defense: MTB produces catalase-peroxidase and other enzymes to neutralize reactive oxygen species (ROS) generated by the oxidative burst.
- Apoptosis inhibition: MTB inhibits macrophage apoptosis, preventing an early bacterial killing pathway; this paradoxically aids persistence.
- Nutrient scavenging: Once inside the phagosome, MTB can access host lipids and fatty acids for nutrition.
Over subsequent days, monocyte-derived macrophages, interstitial macrophages, and neutrophils are recruited to the site, but these too fail to control MTB replication. The interval to adaptive immunity is 2–8 weeks, during which MTB grows relatively unhindered.
Some individuals — particularly those with high-level innate responses, including robust metabolic responses to free fatty acids and MTB-specific antibody responses — may sterilize the infection entirely before adaptive immunity develops.
4. The Adaptive Immune Response
Infected dendritic cells travel to draining lymph nodes, where they present MTB antigens (via MHC class I and II) to naive T cells. Key events:
| Cell | Role |
|---|---|
| CD4⁺ Th1 cells | Secrete IFN-γ and TNF-α → activate macrophages for killing |
| CD4⁺ Th17 cells | Recruit neutrophils via IL-17 |
| CD8⁺ CTLs | Lyse infected macrophages via perforin/granzymes |
| B cells | Antibody production (role still being defined) |
| Regulatory T cells (Tregs) | Dampen excessive inflammation but may impair bacterial killing |
IFN-γ is the pivotal cytokine — it drives macrophage activation, induces nitric oxide synthase (iNOS), and promotes autophagy. Patients with inborn errors of IFN-γ signaling or those who develop IFN-γ–neutralizing antibodies are at exceptionally high risk for disseminated disease.
TNF-α is equally critical: it is required for granuloma formation and maintenance. This explains why patients on TNF-α inhibitors (e.g., infliximab, adalimumab) face markedly elevated TB risk — they cannot maintain organized granulomas.
The adaptive response takes 2–8 weeks to develop, during which tuberculin skin test (TST) and IGRA results convert from negative to positive (tuberculin conversion).
5. Granuloma Formation — The Hallmark
The granuloma is the pathological and immunological centerpiece of TB.
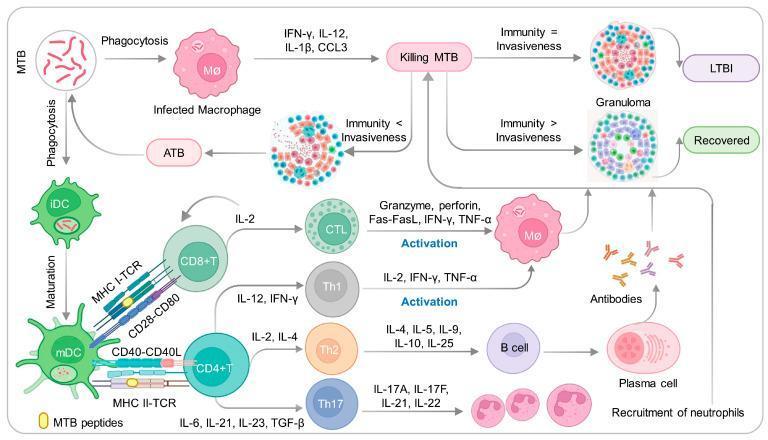
Once antigen-specific T cells return to the lung, they coordinate the formation of an organized collection of:
- Epithelioid macrophages (transformed, activated macrophages) at the center
- Multinucleated Langhans giant cells (fused macrophages)
- Foamy (lipid-laden) macrophages
- CD4⁺ T cells at the periphery
- CD8⁺ T cells and B cells at outer layers, sometimes forming lymphoid follicle-like structures
The granuloma serves to wall off and contain the infection. In the classic TB granuloma, the center undergoes caseous necrosis — the tissue takes on a cheese-like appearance due to a combination of ischemia, cytotoxic T-cell activity, and TNF-mediated cell death. This environment is acidic, hypoxic, and nutrient-poor, which suppresses further bacterial replication.
Granuloma dynamics are not static: even within a single individual, different granulomas can achieve different degrees of bacterial control. Some maintain sterile containment; others harbor viable bacilli.
How granulomas can be a double-edged sword:
- They prevent progressive disease by constraining bacteria
- They may enhance bacterial spread between aggregated macrophages
- They create an immunosuppressive microenvironment
- They keep CD4⁺ T cells at the periphery, physically separated from infected macrophages in the center

6. Spectrum of Outcomes After Initial Infection
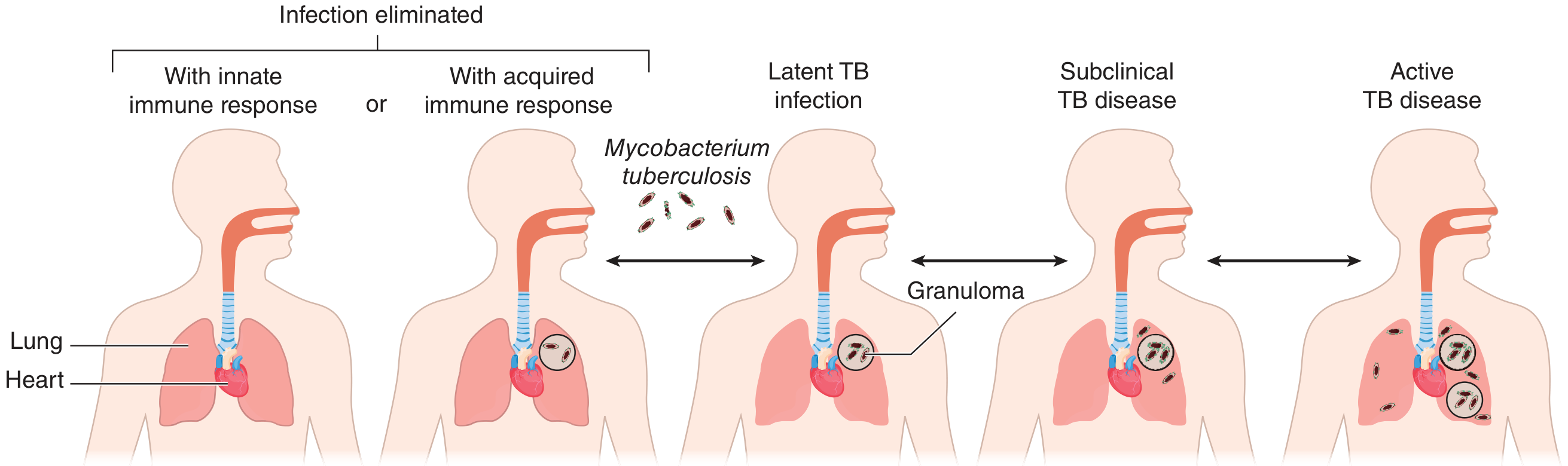
| Outcome | Frequency | Mechanism |
|---|---|---|
| Bacterial elimination | Rare | Robust innate response sterilizes before adaptive response needed |
| Latent TB infection (LTBI) | ~90% of infected | Adaptive immune response controls but does not eradicate |
| Progressive primary TB | ~5–10% | Immune control fails during initial infection (common in children, elderly, immunocompromised) |
| Reactivation TB | ~5–10% lifetime | Immunosuppression (HIV, age, malnutrition, TNF-inhibitors) disrupts granuloma integrity |
| Subclinical disease | Emerging concept | Detectable bacterial replication without symptoms |
7. Latent TB Infection (LTBI)
After the immune response controls primary infection, MTB enters a state of dormancy. The bacilli persist within granulomas, metabolically suppressed by the hostile microenvironment. The Ghon complex (calcified parenchymal focus + calcified hilar lymph node = Ranke complex) may be visible on chest radiography as radiographic evidence of this contained infection.
The TST and IGRA remain positive indefinitely as markers of immune memory. Approximately 5–10% of latently infected immunocompetent individuals reactivate TB at some point in their lifetime; this risk jumps to ~10% per year in untreated HIV co-infection.
8. Reactivation and Active Disease
When host immunity wanes — due to HIV, diabetes mellitus, malnutrition, aging, immunosuppressive drugs, silicosis, or end-stage renal disease — granulomas break down. Key events in reactivation:
- Granuloma liquefaction: Proteolytic enzymes (including matrix metalloproteinases MMP-1, -3, -8, -9, -12) degrade the granuloma matrix. TNF-α and other cytokines drive MMP expression by macrophages.
- Caseous center liquefies → liquid caseum is an ideal medium for exponential MTB growth (organisms multiply extracellularly, reaching 10⁹/mL)
- Cavity formation: Liquefied caseum drains into a bronchus → cavitation. Cavities expose MTB to atmospheric oxygen, further accelerating growth.
- Bronchogenic spread: Cavity contents are coughed up, spreading bacilli to other lung segments → new foci of disease.
- Hematogenous dissemination: Bacilli can enter the bloodstream → miliary TB (seeding of liver, spleen, bone marrow, meninges, kidneys, adrenals).
9. Tissue Pathology and Lung Damage
The clinical manifestations of pulmonary TB arise from:
- Upper lobe predilection: Reactivation preferentially involves the apical and posterior segments of the upper lobes (higher PO₂ favors MTB growth)
- Cavitation → hemoptysis, bronchiectasis, chronic cough
- Fibrosis → restrictive lung disease
- Lymphadenopathy → compression of airways (especially in children)
- Pleural disease: Hypersensitivity reaction to subpleural caseous foci → exudative lymphocytic effusion
The cytokine storm (TNF-α, IL-1β, IFN-γ, IL-6) contributes to systemic symptoms: fever, night sweats, weight loss, anorexia — the classic "consumption" syndrome.
10. HIV–TB Co-infection
HIV selectively depletes CD4⁺ T cells — the very cells required for granuloma maintenance and macrophage activation. Consequences:
- Failure to form or maintain organized granulomas
- Markedly accelerated progression from LTBI to active TB (up to 10% per year vs. 5–10% lifetime in HIV-negative individuals)
- Atypical presentations (lower-lobe involvement, no cavitation, extrapulmonary disease more common)
- Disseminated/miliary TB far more frequent
Summary Pathway
Inhalation of droplet nuclei (1–5 μm)
↓
Deposition in alveoli
↓
Phagocytosis by alveolar macrophages
↓
Phagolysosome fusion arrested → intracellular survival
↓
Innate immune recruitment (2 weeks)
↓
DC migration to lymph nodes → T cell priming (2–8 weeks)
↓
Antigen-specific T cells return to lung
↓
Granuloma formation (containment = LTBI in ~90%)
↓
[Immune control adequate] → LTBI (dormancy, calcification)
[Immune control fails] → Progressive/Active TB
↓ (reactivation)
Granuloma liquefaction → cavitation → dissemination
Key sources:
- Goldman-Cecil Medicine, 26th ed., Ch. 299 (Pathobiology of Tuberculosis)
- Schwartz's Principles of Surgery, 11th ed., Ch. on Mycobacterial Infections
- Murray & Nadel's Textbook of Respiratory Medicine (Tuberculosis and Lymphatics)
This is a shared conversation. Sign in to Orris to start your own chat.