Elicit document and present history in a case of Burns and perform physical examination. Describe Pathophysiology of Burns.
Reading File
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Searching Images
burn depth classification superficial partial full thickness
Finding Sources
Reading File
Reading File
Loading Image
Reading File
Reading File
I now have comprehensive content from multiple textbooks. Let me compile the complete structured answer.
Burns: History, Physical Examination & Pathophysiology
PART I — HISTORY IN A CASE OF BURNS
A thorough history is essential for assessing burn severity, anticipating complications, and guiding management.
1. Presenting Complaint
- Burn wound with pain, blistering, or skin loss
- Associated symptoms: shortness of breath, hoarseness, altered consciousness
2. History of the Burn Injury (AMPLE + Burn-Specific)
A. Circumstances of the Injury
| Question | Clinical Relevance |
|---|---|
| When did the burn occur? | Time since injury → guide fluid resuscitation timing; delays raise concern for non-accidental injury |
| Where did it occur? | Enclosed space → high risk of inhalation injury + carbon monoxide (CO) poisoning |
| How did it occur? | Mechanism determines burn type and depth (see below) |
| Duration of exposure? | Longer exposure = greater depth |
| Was the patient trapped? | Trapped in a car, room → inhalation injury until proven otherwise |
B. Mechanism of Injury
- Thermal (flame/scald): Most common; scalds predominant in children <5 yrs (kettles, hot drinks, bath water); flame burns more common in adults
- Chemical: Alkali burns penetrate deeper than acid burns; occupational history crucial
- Electrical: Document voltage (high vs. low); risk of rhabdomyolysis, cardiac arrhythmia, internal injury out of proportion to skin wound
- Radiation: Sunburn, arc-welding, radiation therapy
- Contact burns: Common in elderly (falls against radiators)
C. First Aid Administered
- Was the burn cooled? (Cool running water ≥20 min within 1 hour of injury — reduces depth and pain)
- Any creams, toothpaste, or traditional remedies applied? (Can obscure depth assessment and introduce infection)
- Clothing removed? (Retained hot clothing continues to burn)
D. Associated Injuries
- Burns from explosions, house fires, or road traffic crashes may have concurrent fractures, blast injuries, or blunt trauma
- Screen for loss of consciousness (CO poisoning, head injury)
E. Symptoms Suggesting Inhalation Injury
- Hoarseness, voice change, stridor
- Cough, soot in sputum
- Shortness of breath, wheezing
- Headache, confusion (CO poisoning — cherry-red lips are a late sign)
- History of being in a burning building/vehicle
F. Past Medical History
- Epilepsy, cardiovascular disease, diabetes, immunosuppression (alter resuscitation targets and healing)
- Psychiatric illness, substance abuse, suicidal intent (up to 80% of admitted burn patients in some populations have an underlying factor such as epilepsy, alcohol/drug use, or mental illness)
- Previous burns or hospital admissions
G. Medications & Allergies
- Anticoagulants, immunosuppressants, beta-blockers (affect wound healing and haemodynamic response)
- Drug allergies (especially to antibiotics, silver-containing dressings)
H. Tetanus Immunisation Status
- Burns are tetanus-prone wounds
I. Nutritional & Social History
- Pre-injury nutritional status (critical for wound healing)
- Living situation, support network, occupational exposures
J. Safeguarding / Non-Accidental Injury (NAI) Screen
Always screen in children and vulnerable adults. Raise concern if:
- Delay in presentation
- Inconsistent history between caregivers
- Unexpected burn pattern or depth (e.g., stocking/glove distribution in a child — suggests deliberate immersion)
- Other unexplained injuries (bruises, fractures)
- Frequent hospital attendances
PART II — PHYSICAL EXAMINATION IN BURNS
Follow ATLS primary and secondary survey principles.
PRIMARY SURVEY (ABCDE)
A — Airway
- Inspect oropharynx: singed nasal/facial hairs, soot in mouth, mucosal erythema or blistering
- Note hoarseness, stridor, drooling — early intubation if airway compromise is imminent (oedema progresses rapidly)
- Perioral burns alone don't confirm airway injury but mandate pharyngeal inspection
B — Breathing
- Respiratory rate, SpO₂ (note: pulse oximetry is falsely normal in CO poisoning — check ABG + carboxyhaemoglobin)
- Listen for wheeze (chemical pneumonitis from inhaled smoke particles), stridor, or reduced air entry
- Observe chest expansion — full-thickness circumferential chest burns can mechanically restrict rib movement
- Chest auscultation: bronchospasm, crepitations
C — Circulation
- Heart rate, blood pressure, capillary refill time
- IV access: two large-bore cannulae (avoid burned tissue if possible)
- Insert urinary catheter — urine output target: 0.5 mL/kg/hr (adults); 1 mL/kg/hr (children <30 kg)
- Signs of haemodynamic shock in burns >15% TBSA
D — Disability
- Glasgow Coma Scale (GCS), AVPU
- Pupil responses
- Blood glucose
- CO poisoning → confusion, agitation, coma
E — Exposure
- Fully expose the patient to assess all burn surfaces
- Maintain temperature (cover with dry clean sheet — prevent hypothermia, especially in children and elderly)
- Check all body surfaces including perineum, intertriginous areas, back
SECONDARY SURVEY — Burn Assessment
1. Burn Depth Classification
| Degree | Layer Involved | Appearance | Sensation | Blistering | Healing |
|---|---|---|---|---|---|
| Superficial (1st degree) | Epidermis only | Erythema, dry, no blisters | Painful | None | 3–5 days, no scarring |
| Superficial partial-thickness (2nd degree) | Epidermis + superficial papillary dermis | Moist, erythematous, blistered, blanches | Very painful | Present, clear fluid | 10–14 days, minimal scarring |
| Deep partial-thickness (2nd degree) | Epidermis + reticular dermis | Mottled red/white, less wet, may not blanch | Reduced sensation | May be present | >21 days, scarring likely; may need grafting |
| Full-thickness (3rd degree) | All skin layers | Leathery, white/brown/black eschar, dry | Painless (nerve destruction) | None | Cannot self-heal; requires grafting |
| 4th degree | Skin + underlying fascia/muscle/bone | Charred, necrotic | None | None | Major reconstruction |
Superficial burns (1st degree) are not included in TBSA calculations.
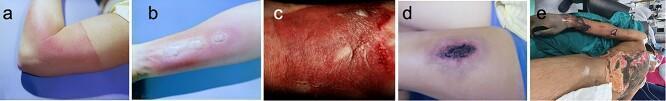
2. Burn Size — Total Body Surface Area (TBSA)
Rule of Nines (Adults):
| Body Part | % TBSA |
|---|---|
| Head & neck | 9% |
| Each arm | 9% |
| Anterior trunk | 18% |
| Posterior trunk | 18% |
| Each leg | 18% |
| Perineum | 1% |
Lund & Browder Chart — preferred in children (adjusts for age-related head:leg ratio differences)
Palmar surface rule: Patient's palm (including fingers) ≈ 1% TBSA — useful for irregular/scattered burns
3. Circumferential Burns
- Check all limbs, chest, and neck for circumferential full-thickness burns
- Signs of compartment syndrome: tense swelling, loss of distal pulses, parasthesia, pain on passive stretch
- Chest: note whether rib expansion is restricted
- Escharotomy indicated for circumferential full-thickness limb or chest burns (incisions placed on lateral aspects of extremities to avoid neurovascular structures)
4. Special Sites
Assess burns involving:
- Face: Airway threat, eyelid/corneal involvement
- Hands/feet: Functional implications — require specialist unit
- Genitalia/perineum: Urinary catheter mandatory
- Major joints: Risk of contracture
- Circumferential limb/chest: Risk of ischaemia/ventilatory compromise
5. Associated Injuries
- Examine for fractures (especially in explosions, falls)
- Check for blast injuries, lacerations
INVESTIGATIONS ORDERED AFTER EXAMINATION
| Investigation | Purpose |
|---|---|
| ABG + carboxyhaemoglobin | Inhalation injury, CO poisoning (SpO₂ misleading) |
| FBC, U&E, creatinine | Baseline, rhabdomyolysis (electrical burns) |
| Blood glucose, coagulation | Baseline |
| CK, myoglobin | Electrical burns → rhabdomyolysis |
| Urine myoglobin | Electrical burns |
| ECG | Electrical burns → arrhythmia |
| CXR | Inhalation injury, baseline |
| Lactate | Cyanide poisoning, shock |
PART III — PATHOPHYSIOLOGY OF BURNS
1. Local Injury — Jackson's Zones of Burn Injury
Jackson (1947) described three concentric zones radiating outward from the point of maximum heat application:
| Zone | Description | Clinical Implication |
|---|---|---|
| Zone of Coagulation (central) | Maximal thermal damage; irreversible cell necrosis; protein coagulation | Full-thickness destruction; cannot be salvaged |
| Zone of Stasis (middle) | Decreased perfusion; cells viable but at risk; capillary endothelial damage with microvascular sludging | Target of resuscitation — can be saved or lost depending on management |
| Zone of Hyperaemia (peripheral) | Increased blood flow, minimal cell injury, inflammatory vasodilation | Heals spontaneously |
Clinical relevance: The zone of stasis can progress to coagulation if resuscitation is inadequate, infection supervenes, or cooling is delayed — this is why 20 minutes of cool water is effective up to 1 hour post-burn.
2. Local Inflammatory Response
Thermal injury triggers an intense local inflammatory cascade:
- Stimulation of pain fibres → release of neuropeptides (substance P, CGRP) → local vasodilation
- Activation of Hageman factor (Factor XII) → initiates the kinin, coagulation, complement, and fibrinolytic cascades
- Arachidonic acid pathway → prostaglandins and leukotrienes → vasodilation + increased vascular permeability
- Complement activation → anaphylatoxins (C3a, C5a) → mast cell degranulation → histamine release → further capillary leak
- Kallikrein–kinin pathway → bradykinin → vasodilatation + increased permeability
Result: Massive capillary leak → oedema formation both locally and systemically (in burns >30% TBSA)
3. Systemic Effects — Burn Shock
In burns involving >15% TBSA (adults), fluid losses become haemodynamically significant:
Haemodynamic sequence:
- Increased vascular permeability → loss of plasma proteins + fluid into interstitium
- Intravascular volume depletion → decreased cardiac preload → decreased cardiac output
- Hypotension → compensatory vasoconstriction → increased afterload → further ↓ cardiac output
- A myocardial depressant factor (MDF) released early post-burn directly impairs myocardial contractility — cardiac output may fall to 30% of baseline within 30 minutes
- Organ hypoperfusion → metabolic acidosis
Fluid kinetics:
- Maximum fluid loss: first 6–8 hours post-burn
- The volume lost is directly proportional to the burn surface area
- Oedema continues to form for 18–24 hours
4. Inhalation Injury
Three distinct mechanisms — can occur alone or together:
| Mechanism | Injury | Cause |
|---|---|---|
| Upper airway injury | Supraglottic oedema, laryngeal oedema | Direct thermal injury from hot gases |
| Lower airway injury | Chemical tracheobronchitis, loss of respiratory epithelium, bronchospasm | Inhaled smoke particles → chemical pneumonitis |
| Metabolic poisoning | Tissue hypoxia | Carbon monoxide (CO) or hydrogen cyanide (HCN) |
Carbon monoxide poisoning:
- CO binds haemoglobin with 250× greater affinity than O₂ → carboxyhaemoglobin (COHb) → impaired O₂ carrying capacity
- CO also competes with O₂ at cytochrome oxidase → disrupts aerobic metabolism → cellular hypoxia even if PaO₂ is normal
- Treatment: High-flow 100% oxygen (reduces CO half-life from 4–5 hours to ~90 minutes)
Cyanide (HCN):
- From combustion of nitrogen-containing polymers (plastics, wool, silk)
- Binds trivalent iron in mitochondrial cytochrome A3 complex → inhibits electron transport → histotoxic hypoxia
- Treatment: hydroxocobalamin (preferred) or sodium thiosulfate
5. Metabolic Response — Hypermetabolism
Burns >30–40% TBSA trigger the most profound hypermetabolic response of any injury:
- Metabolic rate increases to 150–200% of basal (peaks at 2–3 weeks post-burn)
- Driven by: catecholamines, cortisol, glucagon, cytokines (TNF-α, IL-1, IL-6)
- Features: marked catabolism, muscle wasting, impaired wound healing, immunosuppression
- Persistent hypermetabolism can last up to 2 years post-injury
6. Immunological Effects
- Burns produce a biphasic immune response: initial pro-inflammatory phase (SIRS) followed by compensatory anti-inflammatory response (CARS)
- Cell-mediated immunity is significantly reduced
- Depressed neutrophil function, impaired T-cell activity
- Loss of skin barrier → entry point for bacteria
- Sources of infection: burn wound, lungs (pneumonia), central venous catheters, urinary catheters
- Translocation of gut bacteria (impaired gut mucosal barrier) → another infection source in large burns
7. Gastrointestinal Effects
- Inflammatory stimulus + hypovolaemic shock → gut mucosal ischaemia
- ↓ Gut motility, impaired absorption
- Bacterial translocation → systemic sepsis
- Curling's ulcer — acute gastric ulceration in major burns (prevented by enteral feeding and H₂ blockers/PPIs)
- Gut mucosal swelling + peritoneal oedema → abdominal compartment syndrome → splints the diaphragm → ↑ airway pressures
8. Peripheral Circulation and Compartment Syndrome
- Full-thickness burns → collagen coagulation → skin loses elasticity → leathery eschar
- Circumferential full-thickness limb burns: as oedema forms, inelastic eschar acts as a tourniquet → compartment syndrome → limb-threatening ischaemia
- Escharotomy releases this pressure
9. Renal Effects
- Hypovolaemia → ↓ renal perfusion → acute tubular necrosis (ATN)
- In electrical burns: myoglobin + haemoglobin released → tubular precipitation → acute kidney injury
- Adequate fluid resuscitation (urine output target 0.5 mL/kg/hr in adults) is essential
Summary: Pathophysiology at a Glance
BURN INJURY
│
├─► Local: Zones of coagulation / stasis / hyperaemia
│
├─► Inflammatory cascade (Hageman factor, arachidonic acid, complement, kallikrein)
│ └─► Massive capillary leak → oedema → BURN SHOCK (>15% TBSA)
│
├─► Inhalation injury (upper airway, lower airway, CO/CN poisoning)
│
├─► Hypermetabolism + catabolism (peaks week 2–3)
│
├─► Immunosuppression → infection (wound, lung, gut, lines)
│
├─► Gut mucosal ischaemia → ileus, bacterial translocation, Curling's ulcer
│
├─► Circumferential burns → compartment syndrome → escharotomy
│
└─► Renal: ATN from hypovolaemia or myoglobinuria
ABA Burn Center Referral Criteria (Memorise)
-
10% TBSA partial- or full-thickness burns
- Burns involving face, hands, feet, genitalia, perineum, or major joints
- Any full-thickness burn
- Electrical burns (including lightning)
- Chemical burns
- Inhalation injury
- Burns in patients with pre-existing medical conditions
- Burns + concomitant trauma
- Burned children without specialist paediatric facilities
- Burns requiring special social/emotional/rehabilitative intervention
Sources:
- Bailey and Love's Short Practice of Surgery, 28th Ed., Ch. 46 (Burns)
- Schwartz's Principles of Surgery, 11th Ed., Ch. 8 (Burns)
- Mulholland & Greenfield's Surgery, 7th Ed., Ch. 12 (Burns)
- Sabiston Textbook of Surgery (Burns, Paediatric Burns)
Rationale for use of angiotensin converting enzyme (ACE) inhibitors in hypertension
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Finding Sources
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Loading Image
Reading File
Reading File
I now have all the content needed for a thorough, well-structured answer. Let me compile it.
ACE Inhibitors in Hypertension: Rationale for Use
1. Background: Why Target the RAAS?
The renin-angiotensin-aldosterone system (RAAS) is one of the principal physiological regulators of blood pressure, vascular tone, sodium balance, and cardiac and renal structure. In hypertension, excessive RAAS activation drives:
- Systemic vasoconstriction via angiotensin II (Ang II)
- Sodium and water retention via aldosterone
- Cardiac and vascular remodelling (hypertrophy, fibrosis)
- Glomerular hypertension and proteinuria
- Endothelial dysfunction
Blocking this system at the level of ACE is therefore a rational and mechanistically sound antihypertensive strategy.
2. The RAAS Cascade and Site of ACE Inhibitor Action
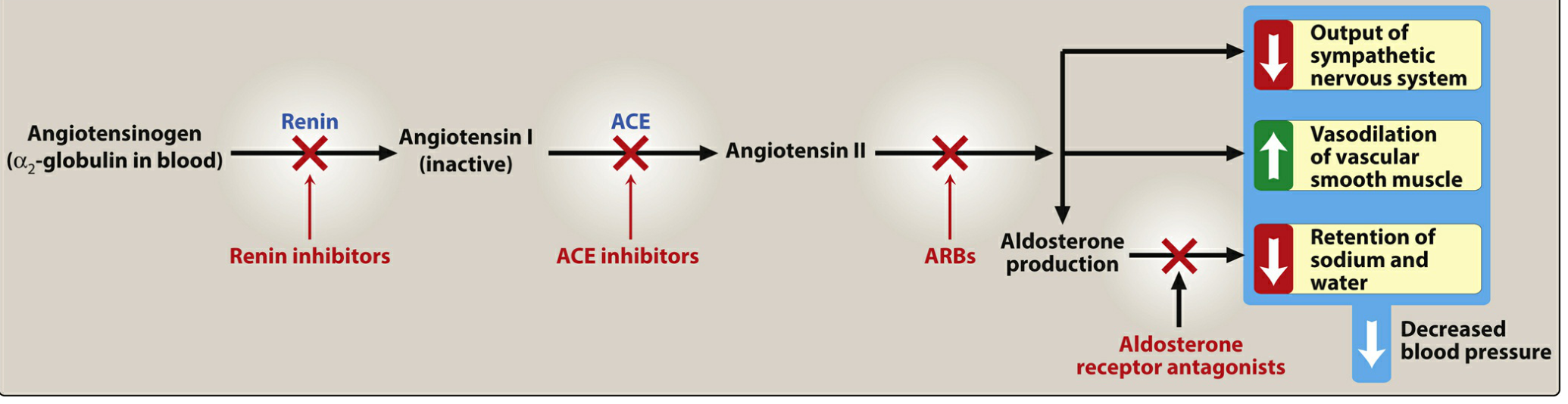
The cascade proceeds as follows:
Angiotensinogen (α₂-globulin)
↓ [Renin — released by juxtaglomerular cells in response to ↓BP, ↓Na⁺, ↑SNS]
Angiotensin I (inactive decapeptide)
↓ [ACE — enzyme in lung endothelium] ← ACE INHIBITORS BLOCK HERE
Angiotensin II (potent vasoconstrictor octapeptide)
↓
├─ AT₁ receptor → Vasoconstriction, ↑aldosterone, ↑SNS activity, cardiac hypertrophy, renal efferent constriction
└─ Aldosterone → ↑Na⁺ + H₂O retention by kidneys
Additionally, ACE (= kininase II) normally degrades bradykinin. ACE inhibitors therefore:
- Block Ang I → Ang II conversion
- Prevent bradykinin breakdown → bradykinin accumulates → stimulates bradykinin B₂ receptors → release of nitric oxide and prostacyclin (potent vasodilators)
3. Mechanisms by Which ACE Inhibitors Lower Blood Pressure
| Mechanism | Effect on BP |
|---|---|
| ↓ Ang II → ↓ vasoconstriction | Arteriolar and venous dilation |
| ↓ Ang II → ↓ aldosterone secretion | ↓ Na⁺ and H₂O retention → ↓ plasma volume → ↓ preload |
| ↑ Bradykinin → ↑ NO + prostacyclin | Additional vasodilation |
| ↓ Sympathetic nervous system activation | ↓ Peripheral vascular resistance |
| ↓ Efferent arteriolar tone in kidney | ↓ Intraglomerular pressure |
Net result: reduction in both peripheral vascular resistance (afterload) and cardiac preload, with sustained blood pressure lowering on chronic treatment.
4. Therapeutic Rationale — Why ACE Inhibitors Are Preferred in Hypertension
4.1 Blood Pressure Lowering Efficacy
- All ACE inhibitors are equally effective in reducing blood pressure at equivalent doses
- Achieve sustained BP reduction with chronic treatment
- Relatively flat dose-response curve; efficacy similar to ARBs
- Less effective as monotherapy in Black patients (low-renin hypertension predominates), but efficacy equalises with combination therapy
4.2 Cardiac Benefits
- Regression of left ventricular hypertrophy (LVH): Ang II directly drives cardiac myocyte hypertrophy and fibrosis via AT₁ receptors and TGF-β; ACE inhibitor use causes measurable regression of LVH
- Post-MI ventricular remodelling: ACE inhibitors improve ventricular remodelling after myocardial infarction, reducing dilatation and progression to systolic heart failure; they are a standard of care post-MI
- Heart failure: ACE inhibitors are first-line agents in heart failure with reduced ejection fraction (HFrEF) — reduce morbidity and mortality; improve endothelial dysfunction and vascular remodelling
- Coronary artery disease risk: ACE inhibitors are first-line drugs in hypertensive patients at increased risk of coronary artery disease
4.3 Renal Benefits — A Key Differentiating Rationale
- Ang II preferentially constricts the efferent arteriole of the glomerulus → raises intraglomerular pressure → promotes proteinuria and glomerulosclerosis
- ACE inhibitors cause efferent arteriolar vasodilation → ↓ intraglomerular pressure → ↓ proteinuria → slow progression of CKD
- Diabetic nephropathy: ACE inhibitors slow progression and decrease albuminuria — a compelling, evidence-based indication independent of blood pressure effect
- Indicated in CKD when urine albumin:creatinine ratio >300 mg/g (Goldman-Cecil)
4.4 Metabolic Neutrality
- Unlike thiazide diuretics and beta-blockers, ACE inhibitors do not cause:
- Hypokalemia
- Hyperglycaemia / insulin resistance
- Dyslipidaemia
- Hyperuricaemia
- They are therefore particularly appropriate in patients with diabetes mellitus and metabolic syndrome
4.5 Endothelial Protection
- Bradykinin accumulation → ↑ NO release → improved endothelial function
- Potential anti-atherosclerotic effects via reduced oxidative stress and inflammation
5. Compelling Indications for ACE Inhibitors in Hypertension
The JNC 7 framework identifies the following conditions where ACE inhibitors have an evidence-based advantage:
| Compelling Indication | ACE Inhibitor Indicated? |
|---|---|
| Heart failure | ✓ Yes |
| Post-myocardial infarction | ✓ Yes |
| High coronary artery disease risk | ✓ Yes |
| Diabetes mellitus | ✓ Yes |
| Chronic kidney disease (CKD) | ✓ Yes |
| Recurrent stroke prevention | ✓ Yes |
Source: Table 27-10, Textbook of Family Medicine 9e (JNC 7 data)
6. Pharmacokinetics — Clinically Relevant Points
| Feature | Detail |
|---|---|
| Route | All are orally bioavailable (as drug or prodrug) |
| Prodrugs | Most (except captopril and lisinopril) require hepatic conversion to active metabolite — prefer captopril/lisinopril in severe hepatic impairment |
| Renal elimination | Nearly all are renally eliminated → dose reduction needed in renal impairment; fosinopril is the exception (dual hepatic/renal) |
| IV form | Only enalaprilat (the active metabolite of enalapril) is available intravenously |
| Onset | Captopril acts fastest (useful in hypertensive urgency) |
Common ACE Inhibitors
| Drug | Trade Name | Note |
|---|---|---|
| Captopril | Generic | Shortest-acting; 3× daily; active drug (no prodrug step) |
| Enalapril | Vasotec | IV form (enalaprilat) available |
| Lisinopril | Prinivil, Zestril | Active drug; once daily |
| Ramipril | Altace | Strong evidence post-MI (HOPE trial) |
| Perindopril | Generic | Used in EUROPA trial (CAD) |
| Fosinopril | Generic | Dual elimination — safe in CKD |
| Benazepril, Quinapril, Trandolapril, Moexipril | Various | Once-daily prodrugs |
7. Adverse Effects — Mechanism and Management
| Adverse Effect | Mechanism | Frequency | Management |
|---|---|---|---|
| Dry persistent cough | ↑ Bradykinin + substance P accumulation in pulmonary tree | 10–15% (more in women, Asian patients) | Switch to ARB |
| Angioedema | ↑ Bradykinin → vascular permeability, extravasation | <1% (more common in Black patients, women, elderly) | Discontinue immediately; ARB is alternative; emergency airway management if severe |
| Hyperkalaemia | ↓ Aldosterone → ↓ K⁺ excretion | Especially in CKD, diabetes, type IV RTA | Monitor K⁺; avoid concurrent K⁺-sparing diuretics |
| First-dose hypotension | Sudden ↓ Ang II in high-renin states | Volume-depleted patients, heart failure | Start low dose; monitor post-first dose |
| Acute rise in creatinine | ↓ Efferent constriction → ↓ GFR (especially bilateral RAS) | Variable | Rise ≤30% above baseline is acceptable; >30% or bilateral renal artery stenosis → discontinue |
| Teratogenicity | ↓ Fetal renal perfusion → oligohydramnios, renal agenesis | — | Absolutely contraindicated in pregnancy (all trimesters) |
8. Contraindications
| Contraindication | Reason |
|---|---|
| Pregnancy | Fetal renal malformations, oligohydramnios, fetal death |
| Bilateral renal artery stenosis | ↓ GFR → acute renal failure |
| History of ACE-inhibitor-induced angioedema | Risk of recurrence; switch to ARB |
| Hyperkalaemia (K⁺ >5.5 mEq/L) | Further K⁺ retention dangerous |
| Concurrent ARB use | Dual RAAS blockade → ↑ hypotension, ↑ renal injury, ↑ hyperkalemia |
| Concurrent aliskiren (in diabetes/CKD) | Same as above |
9. ACE Inhibitors vs Other First-Line Antihypertensive Classes
| Parameter | ACE Inhibitor | ARB | CCB | Thiazide Diuretic |
|---|---|---|---|---|
| Mechanism | Blocks ACE (↓Ang II, ↑bradykinin) | Blocks AT₁ receptor | ↓ Ca²⁺ entry → vasodilation | ↑ Na⁺/H₂O excretion |
| Cough | Yes (10–15%) | No | No | No |
| Angioedema | Yes (<1%) | Rare | No | No |
| K⁺ effect | ↑ K⁺ | ↑ K⁺ | Neutral | ↓ K⁺ |
| Glucose effect | Neutral/beneficial | Neutral | Neutral | ↑ glucose |
| CKD/proteinuria | ✓ First-line | ✓ First-line | Second-line (add to ACEi/ARB) | Less effective alone |
| Black patients | Less effective as monotherapy | Less effective as monotherapy | Very effective | Very effective |
| Post-MI | ✓ Standard of care | Alternative | Some benefit | Less evidence |
| Heart failure | ✓ First-line | Alternative to ACEi | Avoid (HFrEF) | Yes (loop diuretics) |
Note: ACE inhibitors provide equal protection against coronary events, lesser protection against stroke (compared to CCBs), but better protection against heart failure than calcium-channel blockers. - Goldman-Cecil Medicine
10. Summary: The Rationale in Brief
ACE inhibitors are rational and effective antihypertensives because they:
- Directly target the RAAS - the core neurohormonal driver of hypertension and its end-organ consequences
- Lower BP via dual mechanisms - reducing vasoconstriction (↓ Ang II) and enhancing vasodilation (↑ bradykinin → ↑ NO + prostacyclin)
- Reduce cardiac preload AND afterload - lessening myocardial workload
- Reverse hypertensive end-organ damage - LVH regression, improved ventricular remodelling, reduced proteinuria
- Are metabolically neutral - no adverse effects on glucose, lipids, or uric acid
- Carry special benefits in high-risk populations - diabetes, CKD, post-MI, heart failure, CAD
- Are well tolerated - with a predictable, manageable side-effect profile
Sources:
- Lippincott Illustrated Reviews: Pharmacology, Ch. 8 (Antihypertensives)
- Goldman-Cecil Medicine, Ch. 463 (Hypertension)
- Textbook of Family Medicine, 9th Ed., Ch. 27 (Hypertension Management)
- Brenner and Rector's The Kidney, 2-Vol Set (RAAS Physiology)
This is a shared conversation. Sign in to Orris to start your own chat.