A 65-year-old man comes to the emergency department with right upper and lower extremity weakness and slurred speech. The patient went to bed in his usual state of health and woke up with these symptoms. His medical problems include type 2 diabetes mellitus, hypertension, and hyperlipidemia. He takes atorvastatin, metformin, lisinopril, and low-dose aspirin. The patient has smoked a pack of cigarettes daily for the past 40 years. His temperature is 36.7 C (98 F), blood pressure is 160/90 mm Hg, and pulse is 118/min. He is mildly dysarthric with a right lower facial droop and right-sided hemiparesis. A noncontrast head CT is normal. Brain MRI reveals an acute subcortical infarct in the left internal capsule. Six months later, which of the following findings is most likely to be observed in the affected pyramidal tracts of this patient Wallerian degeneration refers to the process that occurs when an axon is damaged, resulting in axonal degeneration and breakdown of the myelin sheath distal to the site of injury. Degeneration of the axon usually begins within a few days after lesion onset. In the peripheral nervous system, Schwann cells sense the axonal degeneration and begin to degrade their myelin and secrete cytokines and chemokines that recruit macrophages. This allows effective clearance of myelin debris which, along with trophic factor secretion by Schwann cells, stimulates formation of a growth cone from the stump of the proximal axon and facilitates nerve regeneration. In the central nervous system, phagocytic macrophages/microglia are recruited more slowly because of the blood-brain barrier. Myelin-producing oligodendrocytes also become inactive or undergo apoptosis and do not assist with phagocytosis. This slows removal of the myelin debris, which can persist for years in the degenerating tracts and suppress axonal growth via myelin-associated inhibitory factors. Astrocytes also release inhibitory molecules and proliferate in the weeks to months following injury, forming a glial scar that acts as a barrier to axon regeneration. (Choice A) Calcification of an ischemic infarction may occur months to years after the event; however, this is a rare occurrence. Brain calcification is most commonly seen with certain neoplasms (eg, oligodendroglioma), infections (eg, neurocysticercosis), and vascular malformations. (Choices B and G) Cytotoxic and vasogenic edema typically occur in the acute setting after ischemic neuronal injury. Cytotoxic edema is a type of intracellular edema caused by impaired function of ATP-dependent sodium/potassium pumps on neuronal cell membranes. In contrast, vasogenic edema is a type of extracellular edema caused by increased vascular permeability due to the effects of pro-inflammatory cytokines. (Choice C) In the first 12-24 hours after ischemic stroke, affected neurons classically shrink and develop intense cytoplasmic eosinophilia (red neurons) with pyknosis of the nucleus. (Choice D) Embolic ischemic strokes have a high risk of undergoing acute hemorrhagic transformation within the first 7 days following the ischemic event. (Choice E) Neutrophilic infiltration progressively increases up to 48 hours after ischemic stroke and then diminishes. Educational objective: Wallerian degeneration refers to the process of axonal degeneration and breakdown of the myelin sheath that occurs distal to a site of injury. Axonal regeneration does not occur in the central nervous system due to the persistence of myelin debris, secretion of neuronal inhibitory factors, and development of dense glial scarring. Convert this UWorld question into ultra high-yield USMLE notes: 1. Diagnosis: 2. Key clue from question stem (1 line): 3. Why correct answer (max 2 lines): 4. Why others wrong (1 line each): 5. Buzzword trigger: 6. Trap / trick tested: 7. One-liner memory hook: Make it extremely concise. No paragraphs.
- (A) Calcification — rare, occurs over years, not the expected finding at 6 months
- (B) Cytotoxic edema — acute phase only (hours); resolves within days
- (C) Red neurons (eosinophilic shrinkage) — seen at 12–24 hours post-infarct, not 6 months
- (D) Hemorrhagic transformation — embolic strokes, acute window (first 7 days)
- (E) Neutrophilic infiltration — peaks at 48 hours, then resolves
- (G) Vasogenic edema — acute/subacute; caused by BBB breakdown from cytokines, resolves early
sub heading of the topic the test maker want to know
| # | Subheading | What They Want You to Know |
|---|---|---|
| 1 | Wallerian Degeneration | Anterograde axonal + myelin breakdown distal to injury site |
| 2 | CNS vs PNS Regeneration | PNS regenerates (Schwann cells); CNS does NOT (oligodendrocytes, glial scar, myelin inhibitors) |
| 3 | Glial Scar Formation | Astrocyte proliferation → physical + chemical barrier to axon regrowth |
| 4 | Timeline of Stroke Pathology | Acute (hrs–days) vs Subacute (days–weeks) vs Chronic (months+) findings |
| 5 | Corticospinal Tract Anatomy | Left internal capsule lesion → right-sided UMN signs (hemiparesis, facial droop) |
Time = Diagnosis — every wrong answer choice maps to a specific time window; knowing the timeline cold eliminates 5 of 6 distractors instantly
| Timeframe | Finding |
|---|---|
| 0–6 hrs | Cytotoxic edema |
| 12–24 hrs | Red neurons |
| 24–48 hrs | Neutrophil infiltration |
| 1–7 days | Hemorrhagic transformation (embolic) |
| Days–weeks | Vasogenic edema |
| Months–years | Wallerian degeneration / glial scar |
| Rare/years | Calcification |
in the above notes i said before diagnosis 1.point give a subheading what test maker is testing from tht question
- (A) Calcification — rare, occurs over years, not the expected finding at 6 months
- (B) Cytotoxic edema — acute phase only (hours); resolves within days
- (C) Red neurons (eosinophilic shrinkage) — seen at 12–24 hours post-infarct, not 6 months
- (D) Hemorrhagic transformation — embolic strokes, acute window (first 7 days)
- (E) Neutrophilic infiltration — peaks at 48 hours, then resolves
- (G) Vasogenic edema — acute/subacute; caused by BBB breakdown from cytokines, resolves early
A 46-year-old woman comes to the emergency department due to sudden-onset, severe occipital headache and nausea. The patient has a history of hypertension and cigarette smoking. Physical examination shows mild disorientation but no focal weakness, sensory loss, or cranial nerve abnormalities. Noncontrast CT scan of the head reveals diffuse hyperdensity in the subarachnoid space. CT angiography reveals a ruptured anterior communicating artery aneurysm. The patient undergoes endovascular coiling of the aneurysm. Four days later, there is sudden deterioration in her level of consciousness and new, right-sided hemiparesis is observed. Repeat CT scan shows no significant changes. Which of the following is the most likely cause of this patient's neurological deterioration? ubarachnoid hemorrhage (SAH) is a life-threatening condition that most commonly results from rupture of a saccular (berry) aneurysm. It classically manifests with the abrupt onset of severe headache (thunderclap headache); confusion, loss of consciousness, and meningismus may also occur. Noncontrast head CT scan reveals blood in the subarachnoid space (between the arachnoid and pia mater). If the patient survives the initial SAH, a number of complications (eg, rebleeding, hydrocephalus, seizures) may occur in the following days to weeks. The most common is arterial vasospasm, which occurs due to the release of vasoconstrictive factors from damaged erythrocytes in the subarachnoid space and the inability of damaged vascular endothelial cells to produce vasodilators (eg, nitric oxide). The resultant unopposed vasoconstriction can cause delayed cerebral ischemia, which manifests as a change in mental status and/or new focal neurological deficits 3-12 days after the SAH. CT scan usually reveals no significant changes despite the severity of the ischemia following vasospasm. Nimodipine, a selective calcium channel blocker, is used after SAH to promote vasodilation and prevent calcium-dependent excitotoxicity, which improves outcomes by reducing the incidence and extent of ischemia due to cerebral vasospasm. (Choices A, B, and E) Although arterial thrombosis, embolism, and vascular inflammation could all cause a change in mental status and focal neurologic deficits, they are not significant complications of SAH. (Choice C) Hydrocephalus may occur hours to days after SAH due to blood breakdown products obstructing the flow of cerebrospinal fluid or clogging the arachnoid villi. Patients develop signs of elevated intracranial pressure (eg, nausea/vomiting), and neuroimaging demonstrates ventricular dilation. (Choice D) Rebleeding after initial SAH carries a high mortality rate and usually occurs early (<24 hours) after the initial bleed. Although symptoms can include a sudden change in level of consciousness and appearance of new neurological deficits, rebleeding is very rare 4 days after a successfully repaired aneurysmal rupture and CT scan would demonstrate newly extravasated blood. Educational objective: The most common complication of aneurysmal subarachnoid hemorrhage is vasospasm, which typically occurs 3-12 days after the initial insult and can cause delayed cerebral ischemia (eg, sudden change in mental status, new focal neurological deficits). The CT scan usually remains unchanged, unlike with rebleeding.
- (A) Arterial thrombosis — not a significant SAH complication
- (B) Embolism — not a significant SAH complication
- (C) Hydrocephalus — occurs hours–days post-SAH; CT would show ventricular dilation
- (D) Rebleeding — peaks <24 hrs; CT would show new blood; rare after successful coiling
- (E) Vascular inflammation — not a recognized SAH complication
See every time don't end your topic and what test maker is testing you. I told you that for your understanding. That's good. Subarachnoid hemorrhage complications. That's it. In the below, if you think it's high yield, I have to know that cerebral hemorrhage complications according to the time, like what is the early complication, what is the delayed complication, like early complication, like hydrocephalus, what presentation, like delayed complication of CHS, so on and so forth. That's it. You know that kind of high yield little table you can do. You should make sense accordingly and do a notenote.
- (A) Arterial thrombosis — not a significant SAH complication
- (B) Embolism — not a significant SAH complication
- (C) Hydrocephalus — occurs hours–days post-SAH; CT would show ventricular dilation
- (D) Rebleeding — peaks <24 hrs; CT would show new blood; rare after successful coiling
- (E) Vascular inflammation — not a recognized SAH complication
⚡ High-Yield Add-On: SAH Complications by Timeline
| Timeframe | Complication | Mechanism | Key Presentation | CT Finding |
|---|---|---|---|---|
| Hours–2 days | Rebleeding | Aneurysm re-ruptures before repair | Sudden LOC, new neuro deficits | New blood on CT |
| Hours–days | Acute Hydrocephalus | Blood blocks CSF flow / clogs arachnoid villi | ↑ICP: nausea, vomiting, altered mental status | Ventricular dilation |
| Days–weeks | Seizures | Cortical irritation from subarachnoid blood | Convulsions | Usually normal |
| Day 3–12 ⭐ | Vasospasm → Delayed Cerebral Ischemia | RBC breakdown products + ↓NO → vasoconstriction | New focal deficits, ↓consciousness | Normal CT |
| Weeks–months | Chronic Hydrocephalus | Fibrosis of arachnoid villi → impaired CSF reabsorption | Classic triad: dementia, gait apraxia, urinary incontinence (NPH-like) | Ventricular dilation |
A 63-year-old man collapses at home after experiencing severe retrosternal chest pain. The emergency medical team finds him with no pulse, and cardiac monitoring shows ventricular fibrillation. Cardiopulmonary resuscitation is performed, and the patient regains spontaneous circulation after several minutes. On arrival at the hospital, he has stable vital signs but does not follow commands or respond to strong stimuli. Which of the following areas is most likely to be affected first by ischemic damage in this patient? A. Caudate nucleus (7%) B. Hippocampus (56%) C. Hypothalamus (7%) D. Substantia nigra (2%) E. Thalamic nuclei (26%) his patient's cardiac arrest, secondary to ventricular fibrillation, caused rapid cessation of cerebral blood flow and global cerebral ischemia. Although cardiac function and circulation were restored after medical intervention, the patient's brain was subjected to ischemia for an unknown length of time, resulting in neurological impairment. Global ischemia lasting longer than 3-5 minutes can cause irreversible damage to neurons. The cells that are typically affected first by hypoxia are the CA1 pyramidal neurons of the hippocampus, which can be damaged by ischemia in as little as 3 minutes. The cerebellar Purkinje cells and neocortex pyramidal neurons are also highly susceptible to damage if ischemia lasts for 5-10 minutes. These neurons are likely vulnerable to ischemia because of their high metabolic demand and unique neurochemical characteristics (eg, inability to repolarize after anoxic depolarization). The hippocampus is located in the medial temporal lobe and plays an essential role in memory, spatial processing, and response inhibition. Selective damage to the hippocampus is common with transient ischemia and can result in an inability to make new memories (anterograde amnesia) and disorientation to place or time. (Choices A, C, D, and E) Although the caudate nucleus, hypothalamus, substantia nigra, and thalamic nuclei are also vulnerable to ischemia if cerebral hypoxia is sustained, they are not as susceptible to damage as the hippocampal CA1 pyramidal neurons. Educational objective: Global cerebral ischemia is typically precipitated by systemic hypotension (eg, cardiac arrest). The cells most susceptible to ischemia are the CA1 pyramidal neurons of the hippocampus. Other areas of the brain that are damaged early in the course of global cerebral ischemia include the cerebellar Purkinje cells and the pyramidal neurons of the neocortex.
- (A) Caudate nucleus — vulnerable but not first
- (C) Hypothalamus — vulnerable but not first
- (D) Substantia nigra — vulnerable but not first
- (E) Thalamic nuclei — vulnerable but requires more sustained ischemia
⚡ High-Yield Add-On: Neuronal Vulnerability in Global Ischemia (by time)
| Ischemia Duration | Region Affected | Cell Type | Clinical Result |
|---|---|---|---|
| ≥3 min ⭐ | Hippocampus (CA1) | Pyramidal neurons | Anterograde amnesia, disorientation |
| 5–10 min | Cerebellum | Purkinje cells | Ataxia, coordination deficits |
| 5–10 min | Neocortex | Pyramidal neurons | Cortical dysfunction, altered consciousness |
| Prolonged | Caudate, Thalamus, Hypothalamus, Substantia nigra | Various | Multi-system neurological deficits |
| Sustained/severe | Watershed zones | Border zone cortex | Proximal limb weakness ("man in barrel" syndrome) |
A 64-year-old woman comes to the emergency department due to headache followed by sudden onset of blindness in her right eye. She was recently admitted to the hospital for fever, pelvic and shoulder girdle pain, and weight loss. This patient's condition is most likely caused by which of the following? emporal arteritis is an inflammatory condition that affects the walls of medium-sized and large arteries. On light microscopic exam, granulomas are seen in the media of the arteries, consisting of mononuclear infiltrates and multinucleated giant cells. Due to the characteristic histologic features, this condition is also called giant cell vasculitis. The changes in the arteries are segmental: abnormal areas are interspersed by segments of normal-appearing arterial wall. Affected segments cause narrowing of the vessel with a decrease in blood supply to the perfused areas. The median age of onset of the temporal arteritis is age 65. Commonly reported symptoms include: Headache: Focal pain over the temple and tenderness on palpation may be present. Sometimes patients have scalp tenderness with hair combing. Superficial temporal artery inflammation may cause pain, nodularity, and thickening found on palpation of the temporal area. Craniofacial pain syndromes: Jaw claudication, tongue claudication, and facial pain may occur. This tends to appear during mastication (chewing) when the blood supply to the corresponding areas does not increase normally due to the narrowing of the arterial lumens. Polymyalgia rheumatica: This related condition occurs in more than half of patients with temporal arteritis. It is characterized by neck, torso, shoulder, and pelvic girdle pain and morning stiffness. Fatigue, fever, and weight loss may also occur. Sudden vision loss is a dreaded complication of temporal arteritis. It may be transient or may result in permanent blindness. Educational objective: Temporal (giant cell) arteritis is an inflammatory condition that affects the walls of medium-sized and large arteries. Polymyalgia rheumatica occurs in more than half of patients with temporal arteritis and is characterized by neck, torso, shoulder, and pelvic girdle pain and morning stiffness. Fatigue, fever and weight loss may also occur. Monocular vision loss is a common complication of temporal arteritis.
- Migraine — no vision loss pattern like this, no PMR, no fever/weight loss
- Atherosclerosis — no systemic inflammatory symptoms, no girdle pain
- MS — younger patients, optic neuritis is painful, no PMR
- Amaurosis fugax (embolic) — transient, no PMR/systemic features
⚡ High-Yield Add-On: GCA — Full Profile
| Feature | Detail |
|---|---|
| Age | >50 (median 65); women > men |
| Vessel affected | Medium/large arteries; superficial temporal, ophthalmic, posterior ciliary arteries |
| Histology | Granulomas in media; multinucleated giant cells + mononuclear infiltrates; segmental involvement |
| Symptoms | Temporal headache, scalp tenderness, jaw claudication, tongue claudication |
| Dreaded complication | Sudden monocular blindness (anterior ischemic optic neuropathy) — irreversible |
| Associated condition | PMR (>50% of GCA): shoulder + pelvic girdle pain, morning stiffness, fever, weight loss |
| Labs | ↑↑ ESR (>50), ↑CRP, normocytic anemia |
| Diagnosis | Temporal artery biopsy (gold standard); skip lesions → long segment needed |
| Treatment | High-dose corticosteroids IMMEDIATELY — do NOT wait for biopsy; vision loss is preventable if treated early |
⚡ GCA vs PMR — Know the Difference
| GCA | PMR | |
|---|---|---|
| Vessel inflammation | ✅ Yes | ❌ No |
| Vision loss risk | ✅ Yes | ❌ No |
| Girdle pain | Sometimes | ✅ Always |
| Biopsy needed | ✅ Yes | ❌ No |
| Treatment | High-dose steroids | Low-dose steroids |
| Overlap | >50% of GCA have PMR | 15% of PMR develop GCA |
whats PMR
- Age >50 (usually 70s), women > men
- Bilateral shoulder girdle pain + stiffness
- Bilateral pelvic girdle pain + stiffness
- Morning stiffness >45 minutes
- Fatigue, fever, weight loss
- Cannot raise arms above head, cannot get up from chair
- Shoulder girdle = shoulders, neck, upper arms
- Pelvic girdle = hips, buttocks, thighs
- ↑↑ ESR, ↑CRP
- Normal CK (muscles are NOT damaged — this differentiates from myositis/myopathy)
| PMR alone | PMR + GCA |
|---|---|
| Girdle pain, stiffness | + headache, jaw claudication, vision loss |
| Low-dose steroids | High-dose steroids STAT |
| No blindness risk | Blindness risk = emergency |
A 65-year-old man is brought to the emergency department due to acute onset of a severe headache and dizziness. He describes it as a throbbing that localizes to the back of his head, with associated vertigo and mild nausea. The patient has a past medical history of type 2 diabetes mellitus, hypertension, chronic obstructive pulmonary disease, and a recent diagnosis of lung adenocarcinoma. He has smoked 1-2 packs of cigarettes daily for the past 30 years. His temperature is 36.7 C (98 F), blood pressure is 170/96 mm Hg, pulse is 80/min, and respirations are 14/min. Noncontrast head CT reveals an acute hemorrhage in the cerebellar vermis without mass effect or midline shift. Which of the following neurologic findings is most likely to be present during this patient's physical examination? he cerebellar vermis modulates axial/truncal posture and coordination via connections with the medial descending motor systems (eg, anterior corticospinal, reticulospinal, vestibulospinal, and tectospinal tracts). Consequently, acute lesions to this region (eg, due to hemorrhage) typically result in truncal ataxia, characterized by a wide-based, unsteady gait. Patients may also develop vertigo and nystagmus due to involvement of the inferior vermis and the flocculonodular lobe (vestibulocerebellum), which modulate balance and ocular movements via connections with the vestibular nuclei and medial longitudinal fasciculus. Other features of cerebellar hemorrhage include nausea and occipital headache. Most cases are caused by hypertensive vasculopathy, but this patient's recent diagnosis of lung adenocarcinoma should also raise concern for hemorrhagic metastasis. (Choice A) Complete ptosis from central lesions may result from injury to the oculomotor nerve fibers within the upper midbrain and is typically associated with contralateral hemiparesis due to adjacent corticospinal tract involvement (eg, Weber syndrome). (Choice B) Oropharyngeal dysphagia is usually associated with lesions in the brainstem due to involvement of cranial nerves IX, X, and XII, and/or their nuclei. Cerebral cortical and subcortical lesions affecting the descending corticobulbar tracts may also lead to oropharyngeal dysphagia. (Choice C) Hemiparesis results from lesions affecting the corticospinal tracts in the cerebral subcortical white matter and brainstem. Cerebellar lesions do not cause pyramidal tract signs or hemiparesis. (Choice D) Limb dysmetria (overshoot/undershoot during targeted movement) is usually associated with lesions affecting the cerebellar hemispheres due to involvement of the lateral descending motor systems (eg, lateral corticospinal tract, rubrospinal tract). Hemispheric cerebellar lesions may also cause tremor during goal-directed activity (intention tremor). (Choice E) Resting tremor is typically associated with parkinsonism due to damage of the substantia nigra and its projections to the striatum. Rubral tremor occurs at rest and during goal-directed activity and is associated with combined lesions to the midbrain, substantia nigra, and superior cerebellar peduncle due to disruption of cerebellothalamic pathways. Educational objective: Acute lesions to the cerebellar vermis typically cause truncal and gait ataxia due to impaired modulation of the medial-descending motor systems. Involvement of the lower vermis and the flocculonodular lobe also causes vertigo/nystagmus due to dysregulation of the vestibular nuclear complex. no unneceessary tables until ultra hy and or unless i prompt
- (A) Complete ptosis — midbrain CN III injury; associated with contralateral hemiparesis (Weber syndrome)
- (B) Oropharyngeal dysphagia — brainstem CN IX, X, XII or corticobulbar tract lesion
- (C) Hemiparesis — corticospinal tract lesion; cerebellar lesions do NOT cause pyramidal signs
- (D) Limb dysmetria — cerebellar hemisphere lesion (lateral cerebellum), NOT vermis
- (E) Resting tremor — substantia nigra/basal ganglia (parkinsonism), not cerebellum
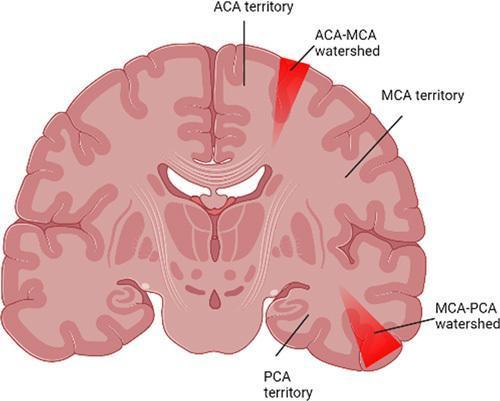
A 58-year-old homeless man is brought to the emergency department after being found unresponsive on a sidewalk. The patient has been hospitalized on several occasions for alcohol and illicit drug intoxication. He also has a history of hypertension but is not adherent with medical therapy. On arrival, the patient is obtunded, febrile, and hypotensive. Physical examination reveals lung crackles and a new blowing diastolic murmur consistent with aortic regurgitation. Appropriate treatment is started, but the patient dies several days later. Autopsy examination shows symmetric, bilateral wedge-shaped strips of necrosis over the cerebral convexity, parallel and a few centimeters lateral to the interhemispheric cerebral fissure. Which of the following is the most likely cause of this patient's cerebral findings? ient with a history of drug use developed fever and new-onset aortic regurgitation, which are suggestive of endocarditis. His pulmonary edema (crackles) and hypotension are due to subsequent cardiogenic shock. On autopsy, the symmetric, bilateral wedge-shaped strips of necrosis over the cerebral convexity are characteristic of global cerebral ischemia. Profound systemic hypotension (eg, cardiogenic shock, cardiopulmonary arrest) diminishes blood supply to the entire brain, resulting in neuron cell death in areas that are vulnerable to hypoxia. This includes brain regions with high metabolic demand (eg, hippocampus, cerebellar Purkinje cells) and areas supplied by the distal-most branches of cerebral arteries (watershed zones). These border zones are located between areas supplied by the anterior, middle, and posterior cerebral arteries and are vulnerable because of low baseline perfusion pressure. The resulting necrosis occurs in bilateral, wedge-shaped strips that can extend from the frontal to occipital lobe and are located parallel and a few centimeters lateral to the interhemispheric fissure. (Choice A) Cerebral amyloid angiopathy typically affects patients over the age of 70 and results in lobar hemorrhages (eg, occipital, parietal) rather than ischemia. Amyloidosis can also cause cardiomyopathy, although patients typically develop right-sided heart failure (eg, edema, ascites) rather than valvular insufficiency. (Choice B) Although infectious endocarditis frequently causes embolization of bacterial vegetations, cardiac embolism would likely cause multiple scattered infarcts in an asymmetric pattern within different major vascular territories (eg, middle and anterior cerebral artery). This patient's symmetric, bilateral infarcts correspond to the cortical watershed regions and are more suggestive of global cerebral ischemia. (Choice D) Charcot-Bouchard aneurysms form as a result of chronic hypertension, and a rupture causes intracerebral hemorrhage typically involving deep brain structures (eg, basal ganglia, cerebellar nuclei, thalamus, pons). (Choice E) Saccular aneurysms, which occur at areas of weakness in cerebral arteries, typically cause subarachnoid hemorrhage after a rupture rather than cerebral ischemia. Patients usually have sudden-onset severe (thunderclap) headache and meningismus. Educational objective: Sustained cerebral hypoperfusion (eg, hypotension, shock) may cause global cerebral ischemia and watershed infarction. These watershed zones are located at the borders between areas perfused by the anterior, middle, and posterior cerebral arteries and typically appear as symmetric, bilateral wedge-shaped strips of necrosis over the cerebral convexity, parallel and adjacent to the interhemispheric fissure. add pic og that watershed areas .
cerebral watershed zones infarction ACA MCA PCA border zones diagram
- (A) Cerebral amyloid angiopathy — age >70; causes lobar hemorrhage, not ischemia
- (B) Cardiac embolism — scattered, asymmetric infarcts in different vascular territories
- (D) Charcot-Bouchard aneurysm rupture — deep hemorrhage (basal ganglia, thalamus, pons); not ischemia
- (E) Saccular aneurysm rupture — SAH; thunderclap headache + meningismus; not ischemic
A 45-year-old man is brought to the emergency department due to new-onset seizure. His family witnessed an episode of blank staring followed by a generalized tonic-clonic convulsion with tongue biting and urinary incontinence. The patient has a past medical history of seasonal asthma and takes no prescription medications. He does not use alcohol or illicit drugs. Vital signs and neurologic examination are normal. Noncontrast head CT and brain MRI are consistent with a diagnosis of cavernous hemangioma. This patient is at greatest risk of developing which of the following complications? avernous hemangiomas are vascular malformations (sporadic or familial) that most commonly occur within the brain parenchyma above the cerebellar tentorium. They consist of clusters of dilated, thin-walled capillaries with little or no intervening nervous tissue. Patients with cavernous hemangiomas often have seizures and may develop progressive neurologic deficits due to mass effect with interruption of the cerebral microcirculation. These lesions also have a tendency to bleed, which can contribute to neurologic symptoms (eg, due to subcortical/cortical tissue irritation) and appear as an irregular hyperdense/bright mass on head CT. (Choice A) Epidural hematomas usually result from rupture of the middle meningeal artery in the setting of head trauma (eg, skull fracture at the pterion). (Choice C) Cavernous hemangiomas increase the risk of hemorrhagic as opposed to ischemic stroke. Ischemic stroke typically occurs in older individuals with cardiovascular risk factors (eg, diabetes mellitus, hypertension, hyperlipidemia, smoking history) due to vessel occlusion caused by thrombosis or cardioembolism. (Choice D) Migraine headache is a primary headache disorder commonly seen in young women. Although migraine headaches can have a secondary cause, they have not been linked specifically to cavernous hemangiomas. (Choice E) Subarachnoid hemorrhage is usually caused by ruptured saccular aneurysms and is an infrequent consequence of bleeding vascular malformations. (Choice F) Subdural hematoma results from rupture of the bridging veins in the subdural space (between the dura and arachnoid membranes) and is most commonly caused by head trauma. Vascular malformations do not typically result in subdural hematomas. Educational objective: Cavernous hemangiomas are vascular malformations that occur most commonly within the brain parenchyma; they carry an increased risk of intracerebral hemorrhage and seizure.
- (A) Epidural hematoma — middle meningeal artery rupture from trauma/skull fracture; not from vascular malformations
- (C) Ischemic stroke — requires atherosclerosis/cardioembolism; cavernous hemangiomas cause hemorrhagic, NOT ischemic stroke
- (D) Migraine — primary headache disorder; not linked to cavernous hemangiomas
- (E) Subarachnoid hemorrhage — ruptured saccular aneurysm; rare with vascular malformations
- (F) Subdural hematoma — bridging vein rupture from trauma; not from cavernous hemangiomas
⚡ High-Yield Add-On: Vascular Malformations Compared
| Type | Structure | Bleed Risk | Presentation | Location |
|---|---|---|---|---|
| Cavernous hemangioma | Dilated thin-walled capillaries; no intervening neural tissue | ✅ High (parenchymal) | Seizures, hemorrhagic stroke | Supratentorial parenchyma |
| AVM (arteriovenous malformation) | Artery-to-vein shunt; no capillary bed | ✅✅ High | Hemorrhage, seizures, headache | Any; often cortical |
| Saccular (berry) aneurysm | Outpouching at vessel bifurcation | ✅✅✅ | Thunderclap headache, SAH | Circle of Willis |
| Epidural hematoma | Middle meningeal artery rupture | Trauma only | Lucid interval → herniation | Between skull & dura |
| Subdural hematoma | Bridging vein rupture | Trauma/elderly | Slow or acute decline | Dura-arachnoid space |
A 60-year-old professor is brought to the emergency department after developing right-sided weakness and numbness during a lecture an hour ago. He also has nausea and confusion. The patient smoked a pack of cigarettes daily for 20 years but quit 10 years ago. He does not drink alcohol. His father died of myocardial infarction at age 70. Blood pressure is 190/100 mm Hg, and pulse is 60 /min. On physical examination, there are no signs of head trauma. There is diminished right-sided strength (1/5) and an upgoing plantar reflex. Brain CT without contrast is obtained immediately. The image is shown below. Explanation Charcot-Bouchard aneurysms Saccular (berry) aneurysms Associated conditions Hypertension ADPKD, Ehlers-Danlos syndrome, hypertension Location Basal ganglia Cerebellum Thalamus Pons Circle of Willis Size <1 mm Variable, 2-25 mm Result of rupture Intracerebral hemorrhage Subarachnoid hemorrhage Symptoms of rupture Progressive neurologic deficits Headache may follow Sudden severe headache Focal neurologic deficits uncommon ADPKD = autosomal dominant polycystic kidney disease. This patient's brain CT is diagnostic for acute hypertensive intracerebral hemorrhage of the thalamus. Hypertensive vasculopathy involving the small penetrating branches of the cerebral arteries is the most common cause of deep intracranial hemorrhage. Chronic hypertension can lead to progressive arteriolar hyalinization and fibrinoid necrosis, weakening the vessel wall and predisposing to the formation of Charcot-Bouchard aneurysms. Aneurysmal rupture may ultimately lead to hemorrhage within the brain. The most common sites of intracranial hemorrhage are deep brain structures, such as the basal ganglia (putamen), cerebellar nuclei, thalamus, and pons. Acute neurologic deficits can vary depending on hemorrhage location. This patient's contralateral weakness/numbness and extensor plantar response is consistent with thalamic hemorrhage with edema/mass effect on the internal capsule. Symptoms may worsen within minutes to hours and can be associated with headache, nausea/vomiting, or decreased level of consciousness as the hematoma expands. Acute bleeding appears as hyperattenuated, or bright, lesions on noncontrast head CT. (Choices A) Arteriovenous malformations are the most common cause of intracranial hemorrhage in children. This patient's older age makes the diagnosis less likely. (Choices B and C) Carotid artery atherosclerosis and cardiac embolism are 2 common causes of ischemic stroke, which can present similar to hemorrhagic stroke; however, this patient's CT shows intracranial hemorrhage (not ischemia). Spontaneous hemorrhagic transformation of ischemic stroke would be rare within hours of symptom onset. (Choice D) Cerebral amyloid angiopathy is the most common cause of lobar/cortical parenchymal hemorrhage, particularly in older adults. This patient's deep intracranial hemorrhage makes hypertensive vasculopathy more likely. (Choice F) Hypertensive encephalopathy is characterized by a progressive headache and nausea/vomiting followed by nonlocalizing neurologic symptoms (eg, confusion). This patient's focal neurologic deficits and evidence of hemorrhage on CT exclude this diagnosis. (Choice G) Hypoxic encephalopathy is caused by decreased oxygenation of the blood (eg, due to cardiac arrest). It most often presents with decreased consciousness (eg, coma or vegetative state); focal neurologic deficits and intracranial hemorrhage would not be seen. (Choice H) Intracranial vasculitis can lead to intraparenchymal hemorrhage but is a far less common etiology compared to hypertensive vasculopathy. (Choice I) Saccular aneurysm rupture is typically associated with subarachnoid hemorrhage as opposed to intraparenchymal hematoma. Patients usually have an abrupt onset of severe headache (thunderclap headache), and focal neurologic deficits are less common. On CT, subarachnoid hemorrhage appears as hyperattenuation of the sulci and basal cisterns. Educational objective: Deep intraparenchymal hemorrhage is most commonly caused by hypertensive vasculopathy of the small penetrating branches of the cerebral arteries (Charcot-Bouchard aneurysm rupture). This is in contrast to rupture of saccular aneurysms, which typically cause subarachnoid hemorrhage.
- (A) AVM — most common hemorrhage cause in children; less likely at age 60
- (B) Carotid atherosclerosis — ischemic stroke; CT shows hypodensity not hemorrhage
- (C) Cardiac embolism — ischemic stroke; hemorrhagic transformation rare within hours
- (D) Cerebral amyloid angiopathy — lobar/cortical hemorrhage in elderly; NOT deep structures
- (F) Hypertensive encephalopathy — no focal deficits; no hemorrhage on CT
- (G) Hypoxic encephalopathy — diffuse; no focal deficits; no hemorrhage
- (H) Intracranial vasculitis — rare; less common than HTN vasculopathy
- (I) Saccular aneurysm rupture — SAH not parenchymal; thunderclap headache; basal cistern hyperattenuation on CT
⚡ High-Yield Add-On: Charcot-Bouchard vs Saccular — Master Table
| Feature | Charcot-Bouchard | Saccular (Berry) |
|---|---|---|
| Size | <1 mm | 2–25 mm |
| Cause | Chronic HTN | ADPKD, Ehlers-Danlos, HTN |
| Location | Basal ganglia, thalamus, pons, cerebellum | Circle of Willis bifurcations |
| Rupture result | Intracerebral hemorrhage | Subarachnoid hemorrhage |
| CT finding | Deep hyperdense parenchymal bleed | Hyperdense sulci/basal cisterns |
| Presentation | Progressive focal deficits, headache, N/V | Thunderclap headache, meningismus |
⚡ HTN Hemorrhage Locations + Deficits
| Location | Deficit |
|---|---|
| Putamen (most common) | Contralateral hemiparesis, hemisensory loss |
| Thalamus | Contralateral hemisensory loss, gaze palsy |
| Pons | Quadriplegia, pinpoint pupils, coma |
| Cerebellum | Ataxia, vomiting, NO hemiparesis |
| Lobar (amyloid angiopathy) | Variable; cortical signs |
A 70-year-old man is brought to the emergency department due to left-sided weakness and slurred speech upon awakening 30 minutes ago. The patient was asymptomatic when he went to bed last night. Medical history includes type 2 diabetes mellitus, hypertension, and hyperlipidemia. The patient is a former smoker with a 30-pack-year history. Physical examination shows a right gaze preference, left lower facial droop, and hemiplegia and hemisensory loss on the left. CT scan of the brain reveals early infarction in the right middle cerebral artery territory with no acute hemorrhage. The patient is hospitalized for further management. Several hours later, he becomes progressively obtunded with elevated intracranial pressure. A repeat CT scan reveals edema in the region of acute infarction causing mass effect. Which of the following is the primary contributor to the pathogenesis of this patient's raised intracranial pressure? his patient had a right hemispheric ischemic stroke and developed altered mental status and increased intracranial pressure (ICP) secondary to cerebral edema. The fixed volume of the skull necessitates that a significant increase in volume of any component of the CNS (eg, brain tissue, blood, cerebrospinal fluid) will raise ICP. This increased ICP can cause progressive neurologic impairment through direct pressure-induced cell injury or mechanical damage due to brain herniation. Irreversible ischemic neural and glial cell injury initiates a sequence of chemical changes that promote edema: Cytotoxic (ionic) edema begins hours after the ischemic injury due to decreased ATP. The failure of ATP-dependent ion pumps, combined with the release of excitatory amino acids (eg, glutamate), leads to the accumulation of intracellular Na+ and water in neural and glial cells despite an intact blood-brain barrier. Vasogenic edema follows 24-48 hours later, when the release of inflammatory mediators disrupts the tight junctions of the blood-brain barrier, allowing proteins (eg, albumin) and water to enter the interstitial space. This vasogenic phase can cause persistent cerebral edema for weeks after the initial injury. (Choice A) Although a decrease in oncotic pressure can cause peripheral edema (eg, nephrotic syndrome), it is not a significant factor in the development of cerebral edema. Mannitol is used in the treatment of cerebral edema because it increases plasma oncotic pressure and temporarily disrupts the blood-brain barrier, allowing movement of fluid out of the CNS to decrease ICP. (Choice C) Inadequate absorption of cerebrospinal fluid at the arachnoid granulations can cause communicating hydrocephalus. Although this process raises ICP, ventriculomegaly would likely be prominent on CT scan. (Choice D) Increased hydrostatic pressure secondary to central venous thrombosis or hypertensive encephalopathy can cause cerebral edema and increased ICP. However, patients typically have gradually worsening headache before development of neurologic abnormalities. (Choice E) Gliosis is the process of repairing damaged tissue in the CNS by proliferating astrocytes and microglia. This process occurs slowly in the months to years after CNS injury and would not be expected to cause acute changes in ICP. Educational objective: The primary mechanism underlying cytotoxic (early) edema following cerebral ischemia is the accumulation of intracellular Na+ and water due to the failure of ATP-dependent ion transporters in hypoxic cells. Cerebral edema causes progressive neurologic decline by raising intracranial pressure, which can directly damage neural tissue or result in mechanical damage due to brain herniation.
- (A) Decreased oncotic pressure — causes peripheral edema (nephrotic); not significant in cerebral edema
- (C) Inadequate CSF absorption — communicating hydrocephalus; CT would show ventriculomegaly
- (D) Increased hydrostatic pressure — venous thrombosis/hypertensive encephalopathy; gradual headache onset, different context
- (E) Gliosis — occurs months–years after injury; not acute
⚡ High-Yield Add-On: Cytotoxic vs Vasogenic Edema
| Feature | Cytotoxic Edema | Vasogenic Edema |
|---|---|---|
| Onset | Hours after ischemia | 24–48 hrs after ischemia |
| Mechanism | ↓ATP → Na⁺/K⁺ pump fails → intracellular Na⁺/H₂O accumulation | Inflammatory mediators → BBB tight junction disruption → protein + H₂O leak into interstitium |
| BBB | Intact | Disrupted |
| Fluid location | Intracellular | Extracellular (interstitial) |
| Cells involved | Neurons + glia swell | Extracellular space expands |
| CT | Subtle early hypodensity | White matter finger-like hypodensity |
| Treatment | Supportive; osmotherapy (mannitol) | Steroids (vasogenic only — NOT for cytotoxic/ischemic) |
| Duration | Days | Weeks |
A researcher is studying neuronal changes in various physiologic and pathologic states. Microscopic examination of neural tissue obtained from experimental animals reveals neurons with shrunken nuclei and eosinophilic cytoplasm lacking Nissl bodies. Which of the following is the most likely cause of these findings? A. Accumulation of abnormal proteins (5%) B. Age-related physiologic changes (8%) C. Defective lysosomal autophagy (4%) D. Irreversible ischemic injury (61%) E. Transection injury of the nerve fiber (17%) F. Transient neural compression (2%) urons are the major functional unit of the nervous system and are characterized histologically by large, round, centrally located nuclei and basophilic cytoplasmic granules composed of rough endoplasmic reticulum (Nissl bodies). When neurons are injured, they undergo distinct histopathologic changes depending on the type of underlying injury. In response to an acute irreversible injury (eg, severe hypoglycemia, ischemia), neurons begin to display characteristic morphologic changes 12-24 hours after the inciting event, appearing as shrunken cells separated from the surrounding tissue. The nucleus becomes pyknotic (shrunken and basophilic with loss of the nucleolus), and the basophilic Nissl bodies disappear, resulting in cytoplasm that stains deeply eosinophilic (red neurons). In the brain, in contrast to most other tissues, ischemia ultimately results in liquefactive necrosis due to the breakdown of neural architecture by hydrolytic enzymes released from reacting inflammatory cells and damaged neurons. The necrotic neuronal remnants are then phagocytized by microglia, and astrocytes proliferate at the site of the injury to form a glial scar. (Choice A) Several neurodegenerative diseases (eg, Alzheimer disease, frontotemporal lobar degeneration) are characterized by intracellular and/or extracellular accumulation of abnormal proteins in neural tissue. Histologic examination of affected areas demonstrates proteinaceous plaques, tangles, and/or inclusions with reactive gliosis and loss of neurons. (Choice B) Normal aging is associated with progressive atrophy and neuronal loss. However, red neuron changes are not characteristic of age-related physiologic changes. (Choice C) Lysosomal storage diseases (eg, Tay-Sachs, Niemann-Pick) cause the accumulation of improperly digested substances in lysosomes, leading to neural degeneration through free radical damage and apoptosis. Expected histopathologic changes include enlarged neuronal cells with a foamy appearance due to accumulated undigested macromolecules. (Choice E) Extensive axon damage or transection results in an axonal reaction characterized by cell body swelling, movement of the nucleus to the periphery, and dispersion of Nissl bodies. This response occurs due to increased protein and lipid synthesis during axon regeneration. (Choice F) Transient neural compression can lead to a temporary loss of function due to ischemia but would not lead to significant histopathologic changes unless the compression was sustained. Neural compression may occur in the CNS due to increased intracranial pressure or a mass lesion. Educational objective: Neurons that sustain irreversible ischemic injury begin to develop characteristic histopathologic changes 12-24 hours after the inciting event. These changes include shrinkage of the cell body, pyknosis of the nucleus, loss of Nissl bodies, and cytoplasmic eosinophilia (red neurons).
- (A) Abnormal protein accumulation — plaques/tangles/inclusions + reactive gliosis (Alzheimer, FTD); no red neuron
- (B) Age-related changes — progressive atrophy + neuronal loss; no red neuron morphology
- (C) Defective lysosomal autophagy — enlarged foamy neurons with accumulated macromolecules (Tay-Sachs, Niemann-Pick)
- (E) Transection injury — axonal reaction: cell body swells, nucleus moves to periphery, Nissl bodies disperse (not lost); opposite of red neuron
- (F) Transient compression — temporary dysfunction; no significant histopathologic change
⚡ High-Yield Add-On: Neuronal Injury Patterns by Cause
| Injury Type | Cell Body | Nucleus | Nissl Bodies | Cytoplasm | Key Cause |
|---|---|---|---|---|---|
| Red neuron (ischemia) | Shrunken | Pyknotic, no nucleolus | Lost | Deeply eosinophilic | Ischemia, hypoglycemia (12–24 hrs) |
| Central chromatolysis (axon transection) | Swollen | Displaced to periphery | Dispersed (not lost) | Pale | Axon injury → regeneration attempt |
| Lysosomal storage disease | Enlarged, foamy | Normal | Obscured | Vacuolated/foamy | Tay-Sachs, Niemann-Pick |
| Neurodegenerative (protein accumulation) | Variable loss | Variable | Variable | Inclusions/tangles/plaques | Alzheimer, Parkinson, FTD |
| Age-related | Progressive loss | Normal → lost | Normal | Normal | Physiologic aging |
A 55-year-old, right-handed woman is brought to the emergency department due to acute-onset headache and difficulty with vision. En route to the hospital, she becomes unconscious. Medical history is significant for hypertension. Blood pressure is 150/90 mm Hg and pulse is 90/min and regular. CT scan of the head without contrast demonstrates an acute hemorrhage in the left temporal lobe with compression of the anterior medial temporal lobe against the free margin of the tentorium cerebelli. Which of the following cranial nerves is most likely to be compromised in this patient? cranial vault is formed by the cranial bones and divided into compartments by the dural folds (ie, falx cerebri, tentorium cerebelli). Because the cranial vault is rigid, there is minimal space for brain expansion if a rapidly enlarging mass (eg, hemorrhage, tumor) or severe, generalized edema (eg, traumatic brain injury) develops. As a result, portions of the brain may protrude through the openings in the dural folds, often according to the following herniation patterns: Subfalcine: herniation of the cingulate gyrus under the falx cerebri Uncal: herniation of the medial temporal lobe (ie, uncus) through the tentorial notch Tonsillar: herniation of the cerebellar tonsils through the foramen magnum This patient with compression of the anterior medial temporal lobe against the free margin of the tentorium cerebelli has uncal herniation. As uncal herniation progresses, the following may occur: Ipsilateral oculomotor nerve (CN III) compression leads to an ipsilateral fixed and dilated pupil. Paralysis of the oculomotor muscles occurs later and leads to ptosis and a down-and-out position of the ipsilateral eye. Ipsilateral posterior cerebral artery compression leads to contralateral homonymous hemianopsia with macular sparing. Compression of the ipsilateral cerebral peduncle of the midbrain against the tentorium may occur, damaging descending corticospinal tracts and causing contralateral hemiparesis. As the midbrain is pushed toward the contralateral side, the contralateral cerebral peduncle may also become compressed against the tentorium, causing ipsilateral hemiparesis (false localizing sign). Without prompt treatment, further pressure on the midbrain can cause downward brainstem displacement (eg, central herniation) and brainstem hemorrhages (ie, Duret hemorrhages) from stretching and rupture of paramedian branches of the basilar artery. This is usually fatal. (Choices A, B, D, E, F, and G) None of the other cranial nerves become compressed due to uncal herniation. Educational objective: Uncal herniation is a possible complication of an expanding ipsilateral mass lesion (eg, hemorrhage, tumor). The first sign of uncal herniation is a fixed, dilated pupil on the side of the lesion. Contralateral or ipsilateral hemiparesis and contralateral homonymous hemianopsia with macular sparing may also occur.
⚡ High-Yield Add-On: Uncal Herniation — Sequence of Events
| Order | Structure Compressed | Finding |
|---|---|---|
| 1st | CN III (parasympathetic fibers) | Ipsilateral fixed, dilated pupil ⭐ |
| 2nd | CN III (motor fibers) | Ipsilateral ptosis + down-and-out eye |
| 3rd | Posterior cerebral artery | Contralateral homonymous hemianopsia (macular sparing) |
| 4th | Ipsilateral cerebral peduncle | Contralateral hemiparesis |
| 5th | Contralateral cerebral peduncle | Ipsilateral hemiparesis (false localizing sign = Kernohan notch) |
| Fatal | Basilar artery paramedian branches | Duret hemorrhages → brainstem death |
⚡ Herniation Patterns — Quick Reference
| Type | What Herniates | Through | Key Finding |
|---|---|---|---|
| Subfalcine | Cingulate gyrus | Under falx cerebri | ACA compression → leg weakness |
| Uncal | Medial temporal lobe (uncus) | Tentorial notch | CN III palsy, blown pupil |
| Central | Diencephalon downward | Tentorial notch | Bilateral signs, Cheyne-Stokes |
| Tonsillar | Cerebellar tonsils | Foramen magnum | Respiratory arrest (most fatal) |
see tables make only when useful for usmle step 1 no extra knowledge i cant rem,ber everything
- No tables unless the comparison is directly tested on Step 1 and genuinely aids memory
- If a table appears, it will be short, focused, and only contain what the exam actually asks
- No "extra knowledge" padding — only what helps you answer the question and close common traps
- Notes stay tight: 7 points + only essential add-ons
yes
A 75-year-old man is brought to the office for evaluation due to memory loss and increasing confusion over the last 6 months. The patient is unable to manage his finances and occasionally gets lost in his neighborhood, needing to ask for directions. On examination, he can recall only 1 of 3 objects after 5 minutes. The patient performs poorly on cognitive assessment testing. Cranial nerves and gait are normal. T2-weighted MRI reveals multiple white matter abnormalities (hyperintensities) within the left frontal, bilateral parietal, and occipital white matter. Also noted are subcortical gray matter hyperintensities in the bilateral thalamus. Which of the following is the most likely diagnosis? Vascular dementia Pathophysiology Ischemia and/or infarcts due to: Large-vessel atherosclerosis Small-vessel disease (eg, arteriolosclerosis) Cerebral amyloid angiopathy Clinical manifestations Early, prominent executive dysfunction Subtypes: Multi-infarct: often with stepwise decline Strategic infarct: associated with localizing cortical deficits Subcortical vascular encephalopathy: associated with subcortical signs (eg, urinary incontinence, gait disturbances) Imaging findings Cortical infarcts Subcortical lacunar infarcts White matter hyperintensities This patient has had a relatively abrupt cognitive decline with prominent executive dysfunction (eg, managing finances, getting lost). Neuroimaging reveals scattered lesions in the white matter and subcortical gray matter, especially the thalamus. This is consistent with vascular dementia (VaD), which often occurs due to one of the following mechanisms: Atherosclerosis of large vessels (eg, basilar arteries, carotid arteries, circle of Willis): Plaque accumulation narrows the lumen; plaque rupture can lead to thrombosis and thromboembolism, often causing a clinical stroke, resulting in localized neurologic deficits. MRI would reveal a discrete cortical infarct. Small-vessel disease, which includes the development of microatheromas and arteriolosclerosis (ie, thickening and stiffening of arteriole walls): Small-vessel disease can lead to microaneurysm formation, obstruction, or breakage of the vessel walls. It more often impacts subcortical regions (eg, thalamus, basal ganglia) because there is less arteriole collateralization in these regions compared to the cerebral cortex. MRI often reveals signs of multiple small infarcts or microbleeds in these areas. Cerebral amyloid angiopathy: Beta-amyloid deposits in the walls of small to medium cerebral arteries and leads to increased fragility of the vessels. Although the most common manifestation is spontaneous, lobar intracranial hemorrhage, it can also cause multiple small infarcts and present as VaD. (Choice A) Although patients with Alzheimer dementia also have progressive memory loss with functional impairments, this disease often has a more insidious onset than VaD. MRI typically reveals parietotemporal cortical atrophy with no evidence of structural disease. (Choice B) Although brain metastases can result in a rapidly deteriorating mental status, they most often cause focal neurologic findings, which are not seen in this patient, and would be visible on MRI as mass lesions, usually located at the gray-white matter junction. (Choice C) MRI in a patient with multiple sclerosis can reveal hyperintensities corresponding to demyelinating plaques, typically located in the periventricular areas. However, MS typically presents in young women (age <50) with neurologic deficits (eg, blurry vision, diplopia, focal weakness/numbness, bowel/bladder dysfunction) disseminated in time and space. (Choice E) Although Wernicke encephalopathy (due to thiamine deficiency) can present with cognitive deficits, the classic triad consists of oculomotor dysfunction, ataxia, and encephalopathy. Chronic thiamine deficiency results in anterograde and retrograde amnesia, confabulation, and apathy. MRI reveals hyperintensities most commonly affecting the mammillary bodies and periaqueductal gray matter. Educational objective: Vascular dementia often presents with prominent executive dysfunction. MRI typically reveals signs of multiple small infarcts, microbleeds, and areas of hyperintensity in the white matter that represent demyelination or axon loss.
- (A) Alzheimer dementia — insidious onset; MRI shows parietotemporal cortical atrophy, no structural lesions
- (B) Brain metastases — focal neuro deficits; MRI shows mass lesions at gray-white junction
- (C) Multiple sclerosis — young women <50; periventricular plaques; relapsing-remitting neuro deficits
- (E) Wernicke encephalopathy — triad: oculomotor dysfunction + ataxia + encephalopathy; MRI shows mammillary body + periaqueductal gray hyperintensities
A 56-year-old male with history of polycystic kidney disease presented to the emergency room because of sudden onset severe headache. He has never had this type of headache in the past. Examination shows some nuchal rigidity. Neurologic examination is within normal limits. Which of the following is the most likely diagnosis? A. Bacterial meningitis (6%) B. Migraine headaches (0%) C. Intracerebral hemorrhage (6%) D. Subarachnoid hemorrhage (84%) E. Temporal arteritis (1%) F. Cluster headache (1%) lthough the major pathologic process in Autosomal Dominant Polycystic Kidney Disease (ADPKD) is in the kidneys, it is a systemic disorder. Cysts and other anomalies are noted in different organs. Intracranial berry aneurysms arise in the circle of Willis, and when these aneurysms rupture they cause subarachnoid hemorrhage (SAH). The clinical presentation described in this question is suggestive of SAH. Patients complain of a severe headache ("the worst headache of their life"); neck stiffness is usually present or develops in the next 24 hours as blood in the subarachnoid space acts as a meningeal irritant. Papilledema and pupillary dilation may be noted, but there are no focal neurologic findings. CT scan of the brain without contrast is the most commonly used initial imaging study for diagnosing SAH and should be done during the first 24 hours from the onset of symptoms. This is positive in up to 90% of patients. If the CT scan does not show SAH, but the clinical suspicion remains high, a lumbar puncture needs to be performed to evaluate the cerebrospinal fluid for the presence of xanthochromia (blood in the CSF). Thus, presence of the xanthochromia in a clinical scenario suggestive of SAH is the most sensitive test for diagnosing SAH. (Choice A) The classic presentation of meningitis includes fever, headache, neck stiffness, photophobia, nausea, vomiting, and signs of cerebral dysfunction (eg, lethargy, confusion, and coma). Additionally, remember to look for clues leading you to the correct answer. For example, the question writer here mentioned that this patient has ADPKD for a reason, to lead you to SAH. (Choice B) Migraine headache is throbbing or pulsatile. It is initially unilateral and localized in the frontotemporal and ocular area, then progresses posteriorly and becomes diffuse. It typically lasts from several hours to an entire day. (Choice C) Intracerebral hemorrhage manifests with headache, vomiting and sudden onset of focal neurological deficits (hemiplegia, hemianesthesia, cranial nerve deficits, aphasia etc). Focal neurologic manifestations are unusual with SAH. (Choice E) An elderly male with scalp tenderness over the temporal area (or headache), blurred vision and intermittent claudication of jaw is a typical presentation of temporal arteritis. This condition is also accompanied by a very high ESR (erythrocyte sedimentation rate) and many times occurs in the setting of polymyalgia rheumatica. Rapid treatment with corticosteroids is necessary to prevent potential blindness. (Choice F) Cluster headache consists of recurrent brief attacks of sudden, severe, unilateral periorbital pain associated with ipsilateral rhinorrhea, and watering of the eyes. Educational Objective: Intracranial berry aneurysms of the circle of Willis are often seen in patients with ADPKD; when ruptured, they cause subarachnoid hemorrhage that presents with sudden onset of "thunderclap headache".
- (A) Bacterial meningitis — fever + gradual headache + signs of cerebral dysfunction; no ADPKD link
- (B) Migraine — throbbing, unilateral, frontotemporal; no nuchal rigidity; no ADPKD link
- (C) Intracerebral hemorrhage — focal neuro deficits (hemiplegia, aphasia); this patient has none
- (E) Temporal arteritis — elderly + scalp tenderness + jaw claudication + ↑ESR; no ADPKD link
- (F) Cluster headache — recurrent, unilateral periorbital pain + ipsilateral rhinorrhea/lacrimation
- 1st test: Non-contrast CT head (positive in 90% within 24 hrs)
- If CT negative but suspicion high: Lumbar puncture → xanthochromia = most sensitive test for SAH
- Xanthochromia = yellow CSF from RBC breakdown products (bilirubin) — appears 2–4 hrs after bleed, persists ~2 weeks
A 58-year-old man comes to the emergency department due to sudden right-sided weakness. He has no sensory loss, problems speaking/swallowing, or difficulty with balance. The patient was previously told that he has elevated blood pressure, but he does not routinely follow up with his physician. He takes no medications. Examination shows intact cranial nerves and sensory function. The patient has 3/5 muscle strength on the right side. Initial CT scan without contrast reveals no abnormalities. Four weeks later, repeat brain imaging shows a 9-mm, fluid-filled cavitary lesion in the left internal capsule. This patient's condition is most likely caused by which of the following? his patient has pure motor hemiparesis and a small cavitary lesion in the internal capsule, characteristic of a lacunar infarct. This type of stroke affects the small penetrating arterioles that supply the deep brain structures (eg, basal ganglia, pons) and subcortical white matter (eg, internal capsule, corona radiata). Lacunar infarcts are primarily associated with chronic hypertension, which promotes lipohyalinosis, microatheroma formation, and hardening/thickening of the vessel wall (hypertensive arteriolar sclerosis). This leads to progressive narrowing of the arteriolar lumen and predisposes to thrombotic vessel occlusion, which causes characteristic clinical syndromes, depending on the portion of the brain affected: Posterior limb of the internal capsule and/or basal pons - pure motor hemiparesis or ataxia-hemiplegia syndrome (ie, ipsilateral limb ataxia out of proportion to motor deficit) Genu/anterior limb of the internal capsule and/or basal pons - dysarthria-clumsy hand syndrome (ie, dysarthria and dysphagia with clumsiness of one hand) Ventroposterolateral or ventroposteromedial thalamus - pure sensory stroke In the acute setting, CT imaging may not reveal the expected hypodensity of ischemic stroke due to the small infarct size (usually <15 mm). After several weeks, these necrotic lesions turn into cavitary spaces filled with cerebrospinal fluid and surrounded by scar tissue called lacunas. (Choices A and B) Cardiac embolism and carotid artery atherosclerosis usually result in occlusion of medium to large cerebral vessels (eg, middle cerebral artery), causing large-territory ischemic infarctions of the cerebral cortex evident on CT scan as large areas of hypodensity. (Choice D) Hypertensive encephalopathy is characterized by progressive headache and nausea/vomiting followed by nonlocalizing neurologic symptoms (eg, confusion). Cerebral imaging may be normal or show parietooccipital white matter edema. (Choice E) Hypoxic encephalopathy occurs from global interruption of the cerebral blood supply (eg, due to cardiac arrest). It commonly presents with decreased consciousness (eg, coma, vegetative state) as opposed to focal neurologic deficits. Cerebral imaging may show watershed infarcts at the border of perfusion zones between the major cerebral arteries. (Choice F) Saccular aneurysm rupture is associated with subarachnoid hemorrhage. Patients usually have abrupt onset of severe headache, and CT will show hyperattenuation (hemorrhage) in the sulci and basal cisterns. Focal neurologic deficits are uncommon. Educational objective: Lacunar infarcts are small, ischemic infarcts (usually <15 mm) involving the deep brain structures (eg, basal ganglia, pons) and subcortical white matter (eg, internal capsule, corona radiata). They most often occur due to hypertension, which causes hardening/thickening of the vessel wall (hypertensive arteriolar sclerosis), predisposing patients with this condition to thrombotic vessel occlusion.
- (A/B) Cardiac embolism / Carotid atherosclerosis — large vessel occlusion → large cortical territory infarct visible on CT; cortical signs present
- (D) Hypertensive encephalopathy — progressive headache + nonlocalizing confusion; no focal deficits; parietooccipital edema on imaging
- (E) Hypoxic encephalopathy — global ischemia → decreased consciousness; watershed infarcts; no isolated focal deficit
- (F) Saccular aneurysm rupture — SAH; thunderclap headache; basal cistern hyperattenuation; no focal deficits
⚡ Lacunar Syndromes — Step 1 Must Know
| Location | Syndrome |
|---|---|
| Posterior limb internal capsule / basal pons | Pure motor hemiparesis |
| Ventroposterolateral thalamus | Pure sensory stroke |
| Genu internal capsule / basal pons | Dysarthria-clumsy hand |
| Basal pons / internal capsule | Ataxic hemiparesis |
A 75-year-old man is brought to the emergency department due to problems with vision and right-sided hemisensory loss that started an hour ago. While in the emergency department, his symptoms gradually worsen and he develops a headache. Head CT reveals multiple, small lobar hemorrhages of varying ages in the occipital and parietal areas with a medium-size acute bleed in the left parietooccipital lobe. Two years ago, the patient developed sudden right arm weakness; neuroimaging at that time demonstrated a small left frontal lobe hemorrhage. He has no head trauma and does not use anticoagulants. This patient most likely suffers from which of the following? his elderly patient likely has recurrent lobar hemorrhage due to cerebral amyloid angiopathy. Amyloid angiopathy is a consequence of β-amyloid deposition in the walls of small- to medium-sized cerebral arteries, resulting in vessel wall weakening and predisposition to rupture. The disease is not associated with systemic amyloidoses; rather, the amyloidogenic proteins are usually the same as those seen in Alzheimer disease. Amyloid angiopathy is the most common cause of spontaneous lobar hemorrhage, particularly in adults age >60. Hemorrhage tends to be recurrent and most often involves the occipital and parietal lobes. Occipital lobe hemorrhage is typically associated with homonymous hemianopsia; parietal hemorrhages can cause contralateral hemisensory loss. Frontal lobe hemorrhage is less common but may result in contralateral hemiparesis. (Choice A) Brain arteriovenous malformation is the most common cause of intracranial hemorrhage in children and tends to be a single lesion. This patient's older age and multifocal hemorrhages make the diagnosis less likely. (Choices B and C) Carotid artery atherosclerosis and cardiac embolism may cause acute ischemic stroke, which typically presents with sudden neurologic deficits that are maximal at symptom onset. Embolic stroke can present with multiple infarcts involving the cerebral cortex and has a higher likelihood of spontaneous hemorrhagic transformation. However, hemorrhagic transformation would be rare within hours of symptom onset. In addition, this patient's gradually worsening symptoms are more consistent with hemorrhagic stroke. (Choice E) Charcot-Bouchard aneurysm rupture is usually due to chronic hypertension and typically causes hemorrhage involving deep brain structures (eg, basal ganglia, cerebellar nuclei, thalamus, pons). This patient's recurrent lobar hemorrhages make this less likely. (Choice F) Hypertensive encephalopathy is characterized by progressive headache and nausea/vomiting followed by nonlocalizing neurologic symptoms (eg, confusion). This patient's focal neurologic deficits and evidence of hemorrhage on neuroimaging exclude this diagnosis. (Choice G) Hypoxic encephalopathy occurs from global interruption of the cerebral blood supply (eg, due to cardiac arrest). It most often presents with decreased consciousness (eg, coma, vegetative state) as opposed to focal neurologic deficits with lobar hemorrhage. (Choice H) Saccular aneurysm rupture is typically associated with subarachnoid hemorrhage as opposed to intraparenchymal hematoma. Patients usually have an abrupt onset of severe headache (thunderclap headache), and focal neurologic deficits are less common. On CT, subarachnoid hemorrhage appears as hyperattenuation of the sulci and basal cisterns. Educational objective: Cerebral amyloid angiopathy is the most common cause of spontaneous lobar hemorrhage, particularly in the elderly. The most common sites of hemorrhage include the occipital and parietal lobes.
- (A) AVM — children; single lesion; not recurrent multifocal
- (B/C) Carotid atherosclerosis / Cardiac embolism — ischemic stroke; hemorrhagic transformation rare within hours; not recurrent lobar pattern
- (E) Charcot-Bouchard — chronic HTN; deep structures (basal ganglia, thalamus, pons); NOT lobar
- (F) Hypertensive encephalopathy — nonlocalizing; no hemorrhage on CT
- (G) Hypoxic encephalopathy — global; decreased consciousness; no focal lobar hemorrhage
- (H) Saccular aneurysm — SAH; thunderclap headache; basal cisterns on CT
A 65-year-old woman is evaluated for worsening urinary urgency and incontinence. The patient has had no abdominal pain, dysuria, or changes in urinary frequency. On several occasions, she has urinated involuntarily, which she attributes to an inability to walk quickly to the bathroom. The patient feels unsteady while ambulating and walks slowly with small steps. Her family also reports that she has become forgetful and has difficulty planning and executing various activities that she used to perform regularly. Medical history is notable for hypertension. The patient does not use tobacco, alcohol, or illicit drugs. Vital signs are within normal limits. Cardiopulmonary, abdominal, and genitourinary examinations show no abnormalities. Neurological examination shows impaired attention and short-term memory. MRI of the brain reveals chronic white matter changes with no infarction, hemorrhage, or mass lesions. There is diffuse ventriculomegaly with no sulcal enlargement. Which of the following is the most likely underlying cause of this patient's urinary incontinence? his patient with impaired memory and cognition, unsteady gait, and urinary incontinence has ventriculomegaly without sulcal enlargement on MRI, which is characteristic of normal pressure hydrocephalus (NPH). NPH can occur due to decreased absorption of cerebrospinal fluid (CSF) by the arachnoid granulations (eg, fibrosis from previous cerebral inflammation or bleeding) with gradual accumulation of CSF in the ventricular system. The lateral ventricles expand, resulting in normal intracranial pressure and stretching the descending cortical fibers (corona radiata). Interruption of these pathways, along with impairment of cortical and subcortical function, leads to the classic triad of NPH symptoms (dementia, gait disturbance, urinary incontinence). The cerebral cortex (through the pontine micturition center) regulates the micturition reflex, which is an autonomic spinal reflex mediated by the neurons in the sacral spinal cord. In NPH, the lack of cortical inhibition results in detrusor muscle hyperactivity. This leads to urinary urgency, which can be worsened by gait abnormalities that limit a patient's ability to quickly get to a bathroom (urge incontinence). Progressive frontal lobe impairment leads to the inability to recognize urinary urge, uncontrolled urination, and a lack of concern as the disease progresses. (Choice A) Cerebellar dysfunction can cause an ataxic gait, dysarthria, and intention tremors. Memory dysfunction and urinary incontinence are more indicative of cerebral impairment. NPH typically spares cerebellar function. (Choice B) Compression of the reticular formation can occur with brainstem herniation, leading to loss of consciousness and coma. (Choice C) Impaired basal ganglia signaling is characteristic of Parkinson disease, which can result in ataxia with a slow gait. However, cogwheel rigidity and resting tremor would be prominent and dementia and urinary incontinence would not be expected until later in the disease course. (Choice D) Spinal cord damage is a common cause of incontinence with gait impairment. However, spinal cord injury also frequently causes lower extremity weakness and is not associated with cognitive impairment. Educational objective: Normal pressure hydrocephalus occurs due to decreased cerebrospinal fluid absorption, resulting in ventriculomegaly with normal intracranial pressure. This causes stretching of the descending cortical fibers (corona radiata) from the frontal lobe and contributes to the classic triad of symptoms (ie, dementia, gait abnormalities, urinary incontinence). Loss of cortical inhibition of the sacral micturition center leads to urge incontinence.
- (A) Cerebellar dysfunction — ataxia + dysarthria + intention tremor; no dementia or incontinence
- (B) Reticular formation compression — brainstem herniation → coma; not NPH
- (C) Basal ganglia impairment — Parkinson's: cogwheel rigidity + resting tremor; dementia/incontinence late
- (D) Spinal cord damage — lower extremity weakness + incontinence; no cognitive impairment
- Gait (wobbly): Corona radiata stretch → frontal-subcortical motor circuit disruption → magnetic/shuffling gait
- Dementia (wacky): Frontal lobe dysfunction → executive dysfunction, memory loss
- Incontinence (wet): Loss of cortical inhibition → detrusor hyperactivity → urge incontinence