Heart failure
Reading File
Asking for Preferences
Finding Sources
Searching PubMed
"heart failure" classification pathophysiology
Reading File
Finding Sources
Reading File
Reading File
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
I now have everything needed for a thorough, clinician-level overview. Let me compose the response.
Heart Failure: Definition, Types, and Pathophysiology
Definition
Heart failure is a clinical syndrome in which the heart cannot pump blood to adequately meet the metabolic demands of peripheral tissues, or can do so only at elevated filling pressures. It is the common end stage of many forms of chronic heart disease - from ischemic heart disease (IHD), hypertension, and valvular disease, to primary cardiomyopathies - though it can also be precipitated acutely by fluid overload, acute MI, or abrupt valvular dysfunction.
- More than 6.5 million people in the US are affected (~2% of the population)
- ~50% of patients die within 5 years of diagnosis
- Roughly 1 million hospitalizations annually in the US
(Robbins, Cotran & Kumar Pathologic Basis of Disease, p. 481)
Classification by Ejection Fraction
| Type | EF | Dominant Mechanism |
|---|---|---|
| HFrEF (heart failure with reduced EF) | ≤40% | Systolic dysfunction - impaired contractile force |
| HFmrEF (mildly reduced EF) | 41-49% | Mixed |
| HFpEF (heart failure with preserved EF) | ≥50% | Diastolic dysfunction - impaired filling |
HFrEF is caused by ischemic injury, pressure/volume overload, or ventricular dilation. Its pathophysiology is relatively well understood.
HFpEF is caused by LV hypertrophy, myocardial fibrosis, constrictive pericarditis, amyloid deposition, or metabolic injury. It tends to affect older women with obesity and type 2 diabetes. Beyond diastolic dysfunction, its pathophysiology involves vascular stiffness, renal sodium avidity, inflammation from regional adiposity, oxidative stress, impaired nitric oxide signaling, and insulin resistance. (Harrison's Principles of Internal Medicine 22E, 2025)
Compensatory Mechanisms (Initial Adaptations)
When cardiac output falls, three major adaptive responses are engaged:
1. Frank-Starling Mechanism
Increased ventricular filling volumes stretch myocytes, increasing actin-myosin cross-bridge formation and augmenting stroke volume. This is effective short-term but leads to progressive ventricular dilation with sustained volume overload.
2. Neurohormonal Activation
- Sympathetic nervous system (SNS): Norepinephrine release increases heart rate, contractility, and peripheral vascular resistance. Also promotes sodium/water retention.
- RAAS: Renin release triggers angiotensin II formation and aldosterone secretion, promoting sodium and water retention, expanding intravascular volume, and increasing vascular tone.
- Vasopressin (ADH): Retained water further increases preload.
- Endothelin: Potent vasoconstrictor; promotes pulmonary hypertension and RV failure.
- TNF-α and IL-1β: Inflammatory cytokines that can contribute to myocyte injury.
These responses maintain blood pressure and organ perfusion short-term, but become maladaptive - driving excessive vasoconstriction, electrolyte abnormalities, baroreceptor dysfunction, direct myocardial toxicity, and arrhythmias.
3. Cardiac Hypertrophy and Remodeling
- In pressure overload (hypertension, aortic stenosis): sarcomeres assemble in parallel → concentric hypertrophy (thickened wall, normal or reduced cavity)
- In volume overload (regurgitation): sarcomeres assemble in series → eccentric hypertrophy (dilated cavity)
Hypertrophied myocytes are not matched by proportional capillary proliferation, creating relative ischemia and predisposing to dysfunction and cell death.
(Robbins, Cotran & Kumar Pathologic Basis of Disease, p. 482)
Ventricular Remodeling - The Vicious Cycle
This is the key driver of HF progression:
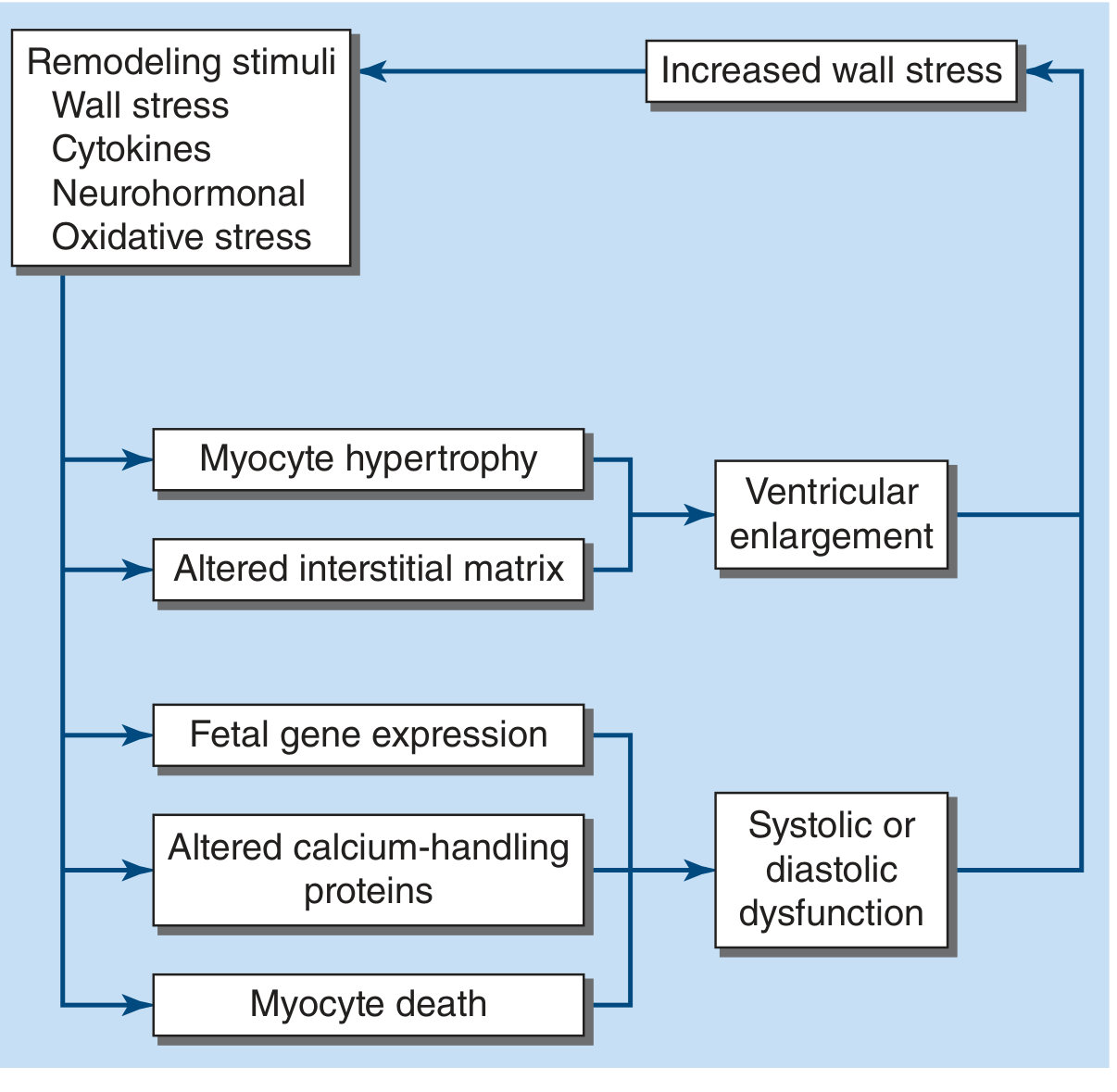
Figure: Remodeling stimuli in heart failure. Hemodynamic stimuli (pressure/volume overload) generate wall stress, cytokines, neurohormonal signals, and oxidative stress. The myocardium responds with myocyte hypertrophy, altered interstitial matrix, fetal gene re-expression, altered calcium-handling proteins, and myocyte death - producing ventricular enlargement and systolic/diastolic dysfunction, which further increases wall stress, completing the cycle. (Harrison's Principles of Internal Medicine 22E)
Cellular changes in remodeling include:
- Abnormal excitation-contraction coupling
- Re-expression of fetal contractile proteins (e.g., β-myosin heavy chain)
- β-Adrenergic receptor desensitization/downregulation
- Impaired cytoskeletal proteins
- Myocyte necrosis, apoptosis, and autophagy
- Interstitial and perivascular fibrosis
- Matrix metalloproteinase activation with ECM degradation
- Ventricular dilation, sphericity, papillary muscle displacement → mitral regurgitation
Counterregulatory (Protective) Hormones
The body also upregulates vasodilatory hormones as a counterbalance:
| Hormone | Source | Actions |
|---|---|---|
| ANP | Atria (stretch-activated) | Natriuresis, vasodilation, inhibits RAAS |
| BNP | Ventricles (stretch/pressure) | Natriuresis, vasodilation, inhibits renin/aldosterone |
| PGE1, PGI2 | Endothelium | Vasodilation |
| Bradykinin | Vascular tissue | Vasodilation |
| Nitric oxide | Endothelium | Vasodilation, inhibits platelet aggregation |
BNP and its precursor NT-proBNP are elevated in HF and used diagnostically for risk stratification. Neprilysin degrades ANP and BNP - hence ARNI therapy (sacubitril/valsartan) inhibits neprilysin to boost these beneficial peptides. (Harrison's Principles of Internal Medicine 22E, p. 2023)
Left-Sided Heart Failure
Causes: IHD, hypertension, aortic and mitral valvular disease, cardiomyopathy.
Pathologic consequences (backward failure - pulmonary congestion):
- Perivascular and interstitial edema → alveolar edema
- Heart failure cells (hemosiderin-laden macrophages) in lung parenchyma - telltale histologic sign
- Pleural effusions (bilateral or right > left; transudative)
Pathologic consequences (forward failure - reduced output):
- Reduced renal perfusion → RAAS activation → salt/water retention → worsens pulmonary edema
- Severe: prerenal azotemia, hypoxic encephalopathy
Clinical features:
- Exertional dyspnea → orthopnea → paroxysmal nocturnal dyspnea (PND) → dyspnea at rest
- Fine bibasal crackles (crepitations)
- S3 gallop (volume overload), S4 (increased wall stiffness)
- Cardiomegaly on CXR; tachycardia
- Progressive dilation → mitral regurgitation → left atrial dilation → atrial fibrillation → thrombus risk
(Robbins, Cotran & Kumar Pathologic Basis of Disease, p. 484)
Right-Sided Heart Failure
Causes:
- Most common: any cause of left-sided HF (elevated pulmonary venous pressure → pulmonary hypertension → RV pressure overload)
- Isolated: cor pulmonale (RV failure from primary pulmonary disease - COPD, PE, pulmonary fibrosis), pulmonic valve disease, tricuspid valve disease
Pathologic consequences (backward failure - systemic venous congestion):
- Liver: Centrolobular congestion and necrosis ("nutmeg liver"), eventually cardiac cirrhosis
- Spleen: Congestive splenomegaly
- Kidneys: Bilateral congestion → more fluid retention than in left HF
- Subcutaneous tissues: Dependent edema (ankles/legs in ambulatory patients; sacral in bedridden)
- Body cavities: Pleural effusions, ascites, pericardial effusion (hydropericardium)
Clinical features:
- Raised JVP with prominent waveforms
- Dependent pitting edema
- Hepatomegaly (tender), ascites
- In isolated right HF: relative absence of pulmonary edema
(Robbins, Cotran & Kumar Pathologic Basis of Disease, p. 484)
Summary: Forward vs. Backward Failure Concept
| Direction | Mechanism | Consequence |
|---|---|---|
| Backward failure | Blood pools in venous capacitance system upstream of the failing ventricle | Left HF → pulmonary congestion; Right HF → systemic venous congestion |
| Forward failure | Reduced cardiac output and tissue perfusion downstream | Reduced organ perfusion, prerenal azotemia, fatigue, hypotension |
Most patients have elements of both.
Common Causes by Category
| Category | Examples |
|---|---|
| Ischemic | Chronic IHD, post-MI |
| Pressure overload | Hypertension, aortic stenosis, coarctation |
| Volume overload | Aortic/mitral regurgitation, high-output states (anemia, thyrotoxicosis, AV fistula) |
| Myocardial disease | Dilated, hypertrophic, restrictive cardiomyopathy |
| Infiltrative | Amyloidosis, sarcoidosis, hemochromatosis |
| Inflammatory | Myocarditis (viral, autoimmune) |
| Arrhythmia | Tachycardia-induced cardiomyopathy |
| Pericardial | Constrictive pericarditis (impairs filling) |
Key sources:
- Robbins, Cotran & Kumar Pathologic Basis of Disease (Heart Failure chapter, pp. 481-485)
- Harrison's Principles of Internal Medicine 22E (2025), Ch. 264 - Mechanisms of Disease Progression
- Goodman & Gilman's Pharmacological Basis of Therapeutics, Ch. 33 - Pathophysiological Mechanisms
- Tintinalli's Emergency Medicine - Pathophysiology of Acute Heart Failure
This is a shared conversation. Sign in to Orris to start your own chat.