myocardial infarction
Reading File
Asking for Preferences
Finding Sources
Searching PubMed
"myocardial infarction"[MeSH Terms] AND management
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Reading File
Reading File
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Reading File
Finding Sources
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Loading Image
Myocardial Infarction - Comprehensive Review
1. Definition & Epidemiology
Myocardial infarction (MI), commonly called a "heart attack," is death of cardiac muscle due to prolonged ischemia. In the United States, roughly 800,000 individuals experience an MI each year - nearly one every 40 seconds - causing almost 400,000 deaths annually. The frequency rises progressively with age and cardiovascular risk factors. About 10% of MIs occur in people under 40, and 45% in people under 65. Female sex is relatively protective before menopause; post-menopausal estrogen decline (with associated increases in cholesterol, blood pressure, and inflammatory markers) erases this advantage, making IHD the leading cause of death in older women. - Robbins, Cotran & Kumar Pathologic Basis of Disease, p. 511
2. Pathogenesis
Coronary Arterial Occlusion (the typical 90% pathway)
The sequence underlying most MIs:
- An atheromatous plaque is eroded or suddenly disrupted by endothelial injury, intraplaque hemorrhage, or mechanical forces - exposing subendothelial collagen and necrotic plaque contents to blood.
- Platelets adhere, aggregate, and are activated, releasing thromboxane A2, ADP, and serotonin - causing further platelet aggregation and vasospasm.
- Coagulation is activated by tissue factor and other mechanisms, adding to the growing thrombus.
- Within minutes, the thrombus can completely occlude the coronary lumen.
When angiography is performed within 4 hours of MI onset, coronary thrombosis is demonstrable in nearly 90% of cases. Notably, these thrombi usually occur at a site that did NOT previously have a critical (>70%) fixed stenosis. - Robbins, Cotran & Kumar, p. 511
Atypical causes (~10% of MIs)
- Vasospasm with or without atherosclerosis (cocaine, ephedrine, catecholamines)
- Embolism (left atrial mural thrombus from AF, endocarditis vegetations, prosthetic material, PFO)
- Small-vessel disease (vasculitis, sickle cell, amyloid deposition)
- MINOCA (myocardial infarction with nonobstructive coronary arteries)
3. Myocardial Response to Ischemia
The first consequence is cessation of aerobic metabolism within seconds, with inadequate high-energy phosphate production and accumulation of lactate. Contractility ceases within ~1 minute.
| Event | Time |
|---|---|
| ATP depletion begins | Seconds |
| Loss of contractility | < 2 minutes |
| ATP reduced to 50% of normal | 10 minutes |
| ATP reduced to 10% of normal | 40 minutes |
| Irreversible cell injury (necrosis) | 20-40 minutes |
| Microvascular injury | > 1 hour |
Only severe ischemia (blood flow ≤10% of normal) lasting 20-40 minutes leads to irreversible necrosis. Progressive loss of viability is complete by 6-12 hours. The benefits of reperfusion are greatest when achieved quickly - the rationale for time-critical reperfusion therapy. - Robbins, Cotran & Kumar, p. 512
Irreversible injury begins in the subendocardial zone (most susceptible - last to receive blood, exposed to highest intramural pressures). With prolonged ischemia, necrosis spreads as a wavefront centripetally toward the epicardium.
4. Infarct Distribution by Coronary Artery
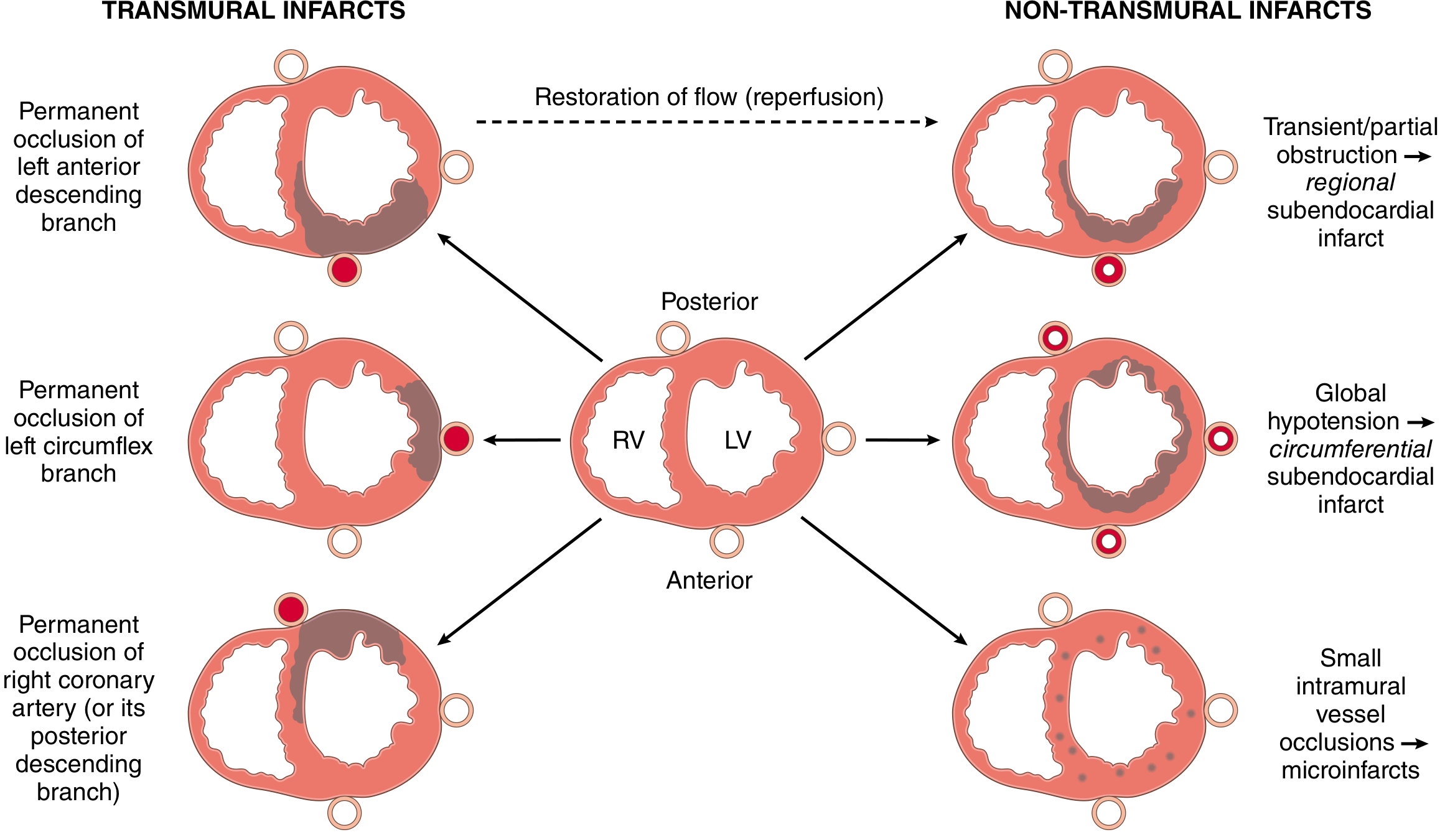
Fig. 12.11 - Robbins, Cotran & Kumar Pathologic Basis of Disease
| Artery | Frequency | Territory |
|---|---|---|
| LAD | 40-50% | Anterior LV wall, apex, anterior 2/3 of septum |
| RCA | 30-40% | Inferior/posterior LV wall, posterior septum, RV free wall |
| LCX | 15-20% | Lateral LV wall (except apex) |
In right-dominant circulation (~80% of people), RCA occlusions can lead to left ventricular damage via the posterior wall. - Robbins, Cotran & Kumar, p. 513
5. Morphological Changes Over Time
| Time | Gross Features | Light Microscopy | Electron Microscopy |
|---|---|---|---|
| 0-0.5 hr | None | None | Myofibrillar relaxation; glycogen loss; mitochondrial swelling |
| 0.5-4 hr | None | Usually none; variable waviness of fibers at border | Sarcolemmal disruption; mitochondrial amorphous densities |
| 4-12 hr | Dark mottling (occasional) | Early coagulative necrosis; edema; hemorrhage | - |
| 12-24 hr | Dark mottling | Coagulative necrosis; nuclear pyknosis; hypereosinophilia; contraction band necrosis; early neutrophilic infiltrate | - |
| 1-3 days | Mottling with yellow-tan center | Coagulative necrosis; brisk neutrophilic infiltrate; loss of nuclei and striations | - |
| 3-7 days | Hyperemic border; central yellow-tan softening | Neutrophil death; macrophage phagocytosis at borders; early granulation tissue | - |
| 7-10 days | Maximally yellow-tan, soft; depressed red-tan margins | Well-developed phagocytosis; granulation tissue at margins | - |
| 10-14 days | Red-gray depressed infarct borders | Established granulation tissue; new vessels; collagen deposition | - |
| 2-8 weeks | Gray-white scar forming from border inward | Fibrosis | - |
| > 2 months | Scarred | Dense collagenous scar | - |
TABLE 12.5 - Robbins, Cotran & Kumar Pathologic Basis of Disease
6. Clinical Features
Patients classically present with:
-
Prolonged chest pain (>30 minutes) - described as crushing, squeezing, or stabbing, often radiating to the left arm, jaw, or shoulder
-
Diaphoresis (profuse sweating)
-
Nausea and vomiting (especially inferior MI with vagal activation)
-
Dyspnea from impaired LV contractility and pulmonary congestion
-
Rapid, weak pulse
-
Silent MI: In up to 25% of patients (especially diabetics with neuropathy), onset is entirely asymptomatic and discovered only by ECG or biomarkers
-
Robbins, Cotran & Kumar, p. 516
7. ECG Changes
STEMI Criteria
In the appropriate clinical context, new ST-segment elevation at the J point in 2 contiguous leads:
-
≥ 0.1 mV in most leads
-
≥ 0.2 mV in V2-V3 in men ≥40 years
-
≥ 0.25 mV in V2-V3 in men <40 years
-
≥ 0.15 mV in V2-V3 in women
-
Frameworks for Internal Medicine, p. 33
Evolution of ECG Changes
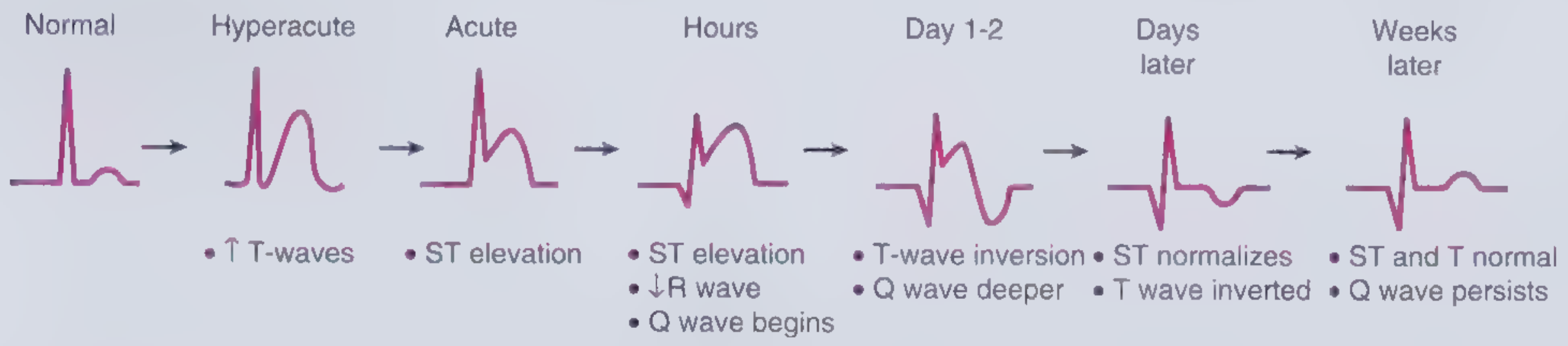
Fig. 2-2 - Frameworks for Internal Medicine
Three mechanisms explain ST elevation in acute MI (Ganong's Physiology):
| Defect in Infarcted Cells | Current Flow | ECG Change |
|---|---|---|
| Rapid repolarization | Out of infarct | ST segment elevation |
| Decreased resting membrane potential | Into infarct | TQ depression (appears as ST elevation) |
| Delayed depolarization | Out of infarct | ST segment elevation |
After days to weeks, Q waves develop (electrically silent dead muscle) and ST changes resolve. Non-Q-wave infarcts tend to be less severe but carry a high risk of subsequent reinfarction. - Ganong's Review of Medical Physiology, p. 534-535
8. Biomarkers
The diagnosis of MI requires elevated cardiac biomarkers plus supporting evidence (ECG changes, symptoms, imaging).
-
Cardiac troponin I and T (cTnI, cTnT) - most clinically useful; proteins that normally regulate calcium-mediated contraction, released when sarcolemmal integrity is lost. Rise within 3-6 hours, peak at 24-48 hours, remain elevated for up to 7-14 days (troponin T) or 5-7 days (troponin I).
-
CK-MB - rises at 4-8 hours, peaks at 18-24 hours, returns to normal at 36-48 hours (useful for detecting reinfarction)
-
An elevated troponin without ST elevation = NSTEMI; with ST elevation = STEMI; elevated troponin without symptoms/ECG changes = type 2 MI or myocardial injury
-
Robbins, Cotran & Kumar, p. 517
9. Classification: STEMI vs. NSTEMI
| Feature | STEMI | NSTEMI/UA |
|---|---|---|
| Occlusion | Complete epicardial occlusion | Partial/transient occlusion |
| ECG | ST elevation ≥ threshold in ≥2 contiguous leads | ST depression, T inversion, or no change |
| Troponin | Elevated | Elevated (NSTEMI) / Normal (UA) |
| Infarct type | Typically transmural | Typically subendocardial |
| Reperfusion urgency | Immediate (primary PCI goal: <90 min) | Urgent but timing varies |
The incidence of STEMI is decreasing (better primary/secondary prevention), while NSTEMI is increasing. - Fuster & Hurst's The Heart, 15th Edition
10. Management
Immediate Goals
-
Identify reperfusion candidates and initiate reperfusion immediately
-
Relieve ischemic pain
-
Recognize and treat hypotension, pulmonary edema, and arrhythmia
-
Mortality is directly related to ischemia time - the benefit of reperfusion is inversely proportional to time to reperfusion
-
Washington Manual of Medical Therapeutics
Upstream Medications
| Drug | Dose | Notes |
|---|---|---|
| Aspirin | 162-325 mg (chewed/crushed) | Rapid platelet inhibition; give to all |
| Clopidogrel | 600 mg load, 75 mg/d | Caution in elderly; 600 mg preferred loading dose |
| Prasugrel | 60 mg load, 10 mg/d | More potent than clopidogrel; avoid if >75 yr, <60 kg, or prior stroke/TIA |
| Ticagrelor | 180 mg load, 90 mg bid | Mortality benefit over clopidogrel; ASA ≤100 mg |
| UFH | 60 units/kg IV bolus, 12 units/kg/h | Give with PCI and thrombolytics (except streptokinase) |
| Enoxaparin (LMWH) | 30 mg IV bolus, then 1 mg/kg SC | Validated in thrombolysis; dose-adjust in elderly/renally impaired |
| Bivalirudin | 0.75 mg/kg IV bolus, 1.75 mg/kg/h | Validated in PCI |
| Nitroglycerin | 0.4 mg SL q5 min x3, then IV 10-200 µg/min | For ongoing chest discomfort; avoid with hypotension |
| Metoprolol | 5 mg IV q2-5 min x3, then 50 mg PO q6h | Reduces reinfarction and VF risk; avoid with HF, bradycardia, shock |
- Washington Manual of Medical Therapeutics
Reperfusion Strategy (STEMI)
- Primary PCI: Preferred if available within 90 minutes of first medical contact - directly opens the culprit artery
- Thrombolytics (fibrinolytics): Used if PCI not available within 120 minutes; then transfer to PCI-capable center
- Calcium channel blockers are not beneficial acutely; short-acting dihydropyridines may increase mortality - Harrison's Principles of Internal Medicine, 22nd Edition
Morphine (analgesic)
- Given 2-4 mg IV, repeated every 5 minutes for pain control
- Caution: may reduce cardiac output by venous pooling; vagotonic effect can cause bradycardia (treat with atropine 0.5 mg IV)
- Harrison's Principles of Internal Medicine, 22nd Edition, p. 2162
11. Complications
Nearly three-quarters of patients experience one or more complications after acute MI. In-hospital mortality is <7% overall (STEMI ~9%, NSTEMI ~6%). One-third of out-of-hospital STEMIs are fatal, usually from arrhythmia within the first hour. - Robbins, Cotran & Kumar, p. 518
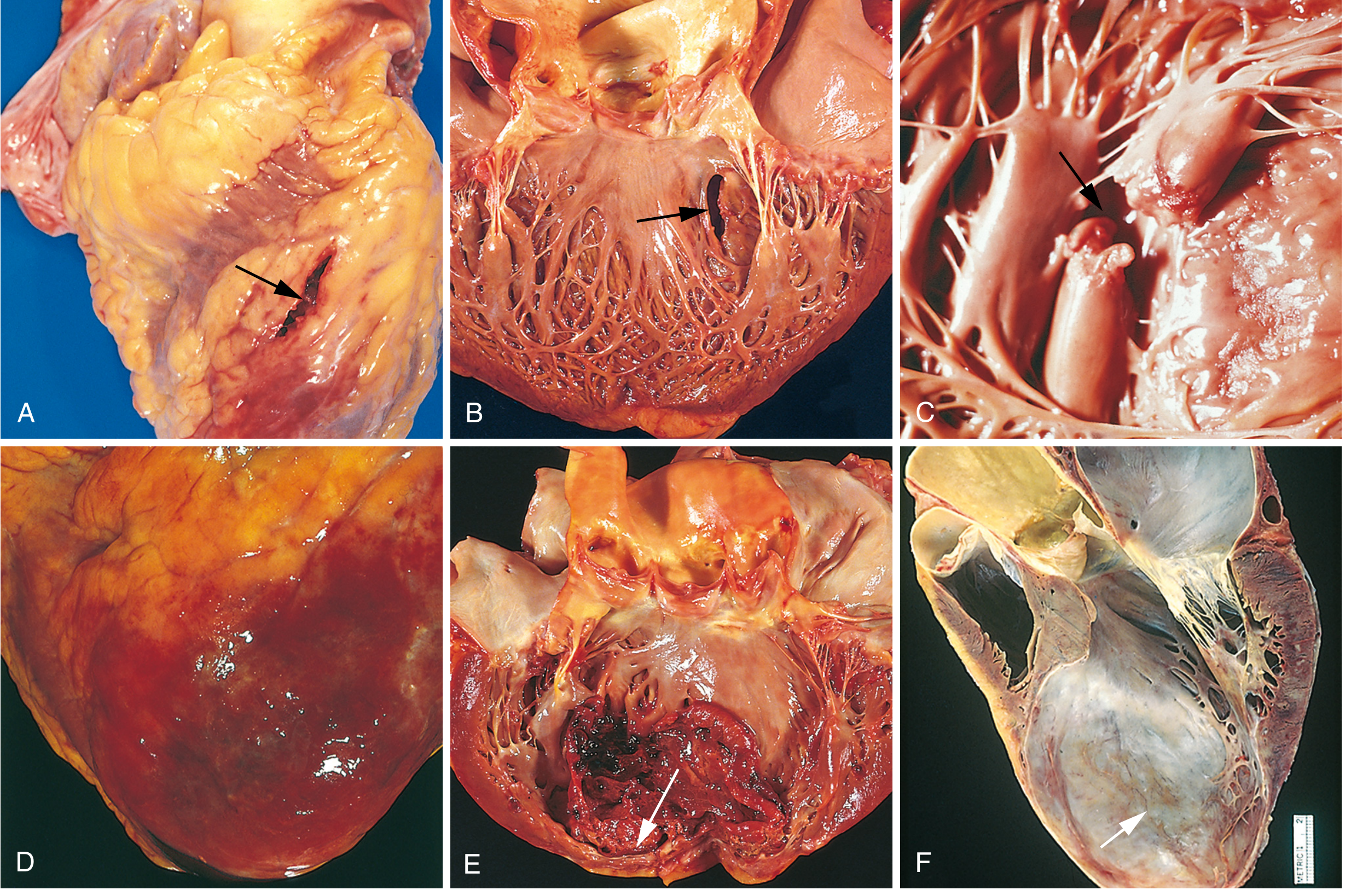
Fig. 12.17 - Robbins, Cotran & Kumar Pathologic Basis of Disease
Complications Summary
| Complication | Details |
|---|---|
| Contractile dysfunction | LV failure proportional to volume of damage; cardiogenic shock in ~10% of transmural MIs (requires ≥40% LV damage) |
| Arrhythmias | ~90% of patients develop rhythm disturbances; VF risk greatest in first hour; includes heart block, VT/VF, supraventricular tachyarrhythmias |
| Myocardial rupture | 1-3% of MIs; most common at 3-7 days (maximal necrosis/softening); free wall rupture → cardiac tamponade (fatal); septal rupture → VSD; papillary muscle rupture → severe MR |
| Papillary muscle dysfunction | Ischemic dysfunction → post-infarct mitral regurgitation |
| Right ventricular infarction | Occurs with RCA occlusions; causes right-sided heart failure, systemic hypotension |
| Pericarditis | Fibrinohemorrhagic; appears 2-3 days after infarct; friction rub; Dressler syndrome (autoimmune) weeks later |
| Mural thrombus / LV thrombus | Risk highest with LAD culprit, reduced EF; LV thrombus in ~1.6% of primary PCI-treated STEMIs (pre-reperfusion era: 15-25%) |
| Infarct expansion / LV aneurysm | Disproportionate thinning and dilation of necrotic wall; can form bulging aneurysm |
| Progressive HF / remodeling | Surviving myocardium hypertrophies; sustained neurohormonal activation leads to dilated cardiomyopathy |
Robbins, Cotran & Kumar, p. 518-519; Textbook of Clinical Echocardiography
12. Secondary Prevention (Post-MI)
Key interventions after discharge:
- Dual antiplatelet therapy (aspirin + P2Y12 inhibitor) for at least 12 months post-ACS
- High-intensity statin (e.g., atorvastatin 40-80 mg)
- ACE inhibitor or ARB (especially with reduced EF or diabetes)
- Beta-blocker (strong evidence in patients with reduced EF; evidence in preserved EF patients is more nuanced - see below)
- Cardiac rehabilitation (home-based or center-based - both shown effective per Cochrane review, 2023)
- Risk factor modification: smoking cessation, BP control, glycemic control, dietary changes
Recent evidence note: A 2025 meta-analysis in European Journal of Preventive Cardiology (PMID 39298680) questioned routine beta-blocker use post-MI in patients without reduced ejection fraction or heart failure, finding uncertain mortality benefit in this preserved-EF group. This is under active debate.
The 2025 ACC/AHA/ACEP Guideline for Management of Acute Coronary Syndromes (PMID 40014670) is the most current practice guideline for ACS management as of 2025.
13. Key Diagnostic Algorithm
Chest Pain / Suspected MI
↓
12-lead ECG within 10 min of arrival
↓
┌─────┴─────┐
STEMI No ST elevation
↓ ↓
Immediate Serial ECGs + Troponins
reperfusion ↓
(primary PCI Elevated Troponin?
<90 min) ↓ ↓
NSTEMI Normal → UA / alternative dx
↓
Risk stratify → early invasive strategy
Key references:
- Robbins, Cotran & Kumar Pathologic Basis of Disease - pathogenesis, morphology, complications
- Ganong's Review of Medical Physiology, 26th Ed. - ECG mechanisms
- Frameworks for Internal Medicine - STEMI/NSTEMI classification, ECG criteria
- Harrison's Principles of Internal Medicine, 22nd Ed. - pain management, reperfusion strategy
- Washington Manual of Medical Therapeutics - medications and protocol
- Fuster & Hurst's The Heart, 15th Ed. - epidemiology, reperfusion
- 2025 ACC/AHA ACS Guideline - current clinical practice
This is a shared conversation. Sign in to Orris to start your own chat.