i need a notes on systemic pathology from guyton and hall
Reading File
Asking for Preferences
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Here are your full textbook-style notes on Systemic Pathology drawn directly from Guyton and Hall Textbook of Medical Physiology:
Systemic Pathology - Guyton and Hall Notes
PART I: CARDIOVASCULAR SYSTEM PATHOLOGY
1. Heart Failure
1.1 Circulatory Dynamics in Heart Failure
Heart failure occurs when the heart cannot pump sufficient blood to meet the body's metabolic demands. Two immediate consequences follow:
- Reduced cardiac output
- Damming of blood in the veins - causing elevated venous pressure
1.2 Acute Effects of Heart Failure
When the heart is suddenly severely damaged (e.g., myocardial infarction), cardiac output falls immediately. In a typical severe case:
- Cardiac output drops from the normal 5 L/min to ~2 L/min (about two-fifths of normal)
- Right atrial pressure rises to +4 mm Hg (blood dams up)
- Symptoms: dizziness/fainting, fatigue, shortness of breath on exertion
Sympathetic Compensation (within seconds):
- Baroreceptor reflex activates; chemoreceptor and CNS ischemic responses also contribute
- Parasympathetic signals to the heart are simultaneously inhibited
- Effects:
- Strengthens damaged ventricular musculature
- Increases heart rate
- Increases venous tone, raising mean systemic filling pressure from ~7 mm Hg to 12-14 mm Hg (nearly 100% above normal)
- Increases venous return
1.3 Compensated vs. Decompensated Heart Failure
Compensated Heart Failure - Sympathetic reflexes and fluid retention restore cardiac output to normal (or near-normal) at rest, but the patient develops:
- Increased venous pressure
- Edema
- Reduced exercise tolerance
Decompensated Heart Failure - When compensation fails:
- Progressive fluid retention further dilates and weakens the heart
- A "vicious cycle" ensues: more fluid retention → more cardiac distension → weaker pumping → more fluid retention
- Ultimately leads to pulmonary edema and death if untreated
Key mechanisms of decompensation:
- Excessive cardiac dilation stretches sarcomeres beyond optimal length, reducing force of contraction
- Edema of the heart muscle reduces efficiency
- Fibrosis replaces functional myocardium
1.4 Unilateral Left Heart Failure
When only the left ventricle fails:
- Left atrial pressure rises markedly
- Pulmonary capillary pressure rises, often causing pulmonary edema
- Right side continues to pump blood into the lungs
- Result: fluid accumulates in the pulmonary interstitium and alveoli
- This is the pathological basis of cardiac asthma (paroxysmal nocturnal dyspnea)
1.5 Low-Output Cardiac Failure
The most common type - reduced cardiac output. Causes:
- Myocardial infarction
- Cardiomyopathy
- Severe valvular disease
1.6 High-Output Heart Failure
Paradoxically, the heart can fail even with supranormal cardiac output when the metabolic demands of the body exceed what even an enhanced output can supply:
- Causes: severe anemia, thyrotoxicosis, beriberi (thiamine deficiency), arteriovenous fistulas
- The heart works harder than normal but still cannot maintain adequate tissue oxygen delivery relative to demand
1.7 Heart Failure with Preserved Ejection Fraction (HFpEF)
- EF is normal (≥50%) but the heart is stiff and cannot fill properly (diastolic dysfunction)
- The ventricle fills at abnormally high pressures
- Results in elevated left atrial and pulmonary venous pressures → pulmonary congestion
- Common in hypertension, diabetes, obesity, and the elderly
- Pathology: myocardial fibrosis, impaired calcium re-uptake in cardiomyocytes, decreased compliance of the LV
1.8 Cardiogenic Shock
Occurs when the heart cannot maintain even the minimum cardiac output for survival:
- Survival rate is often less than 30% even with appropriate medical care
- The vicious cycle: low BP → reduced coronary perfusion → further myocardial damage → even lower BP
- In healthy hearts, this cycle begins at BP <45 mm Hg
- In hearts with already-blocked coronary vessels, the cycle starts at BP 80-90 mm Hg - hence even mild hypotension is dangerous in MI patients
Treatment of Cardiogenic Shock:
- Positive inotropes (increase contractile strength)
- Vasopressors (raise arterial pressure)
- Mechanical circulatory support:
- Intra-aortic balloon pump (IABP) - inflates in diastole (increases coronary flow), deflates in systole (reduces afterload)
- Ventricular assist devices (VADs) - left ventricle pumps blood through a cannula into the aorta
- Revascularization:
- Coronary angioplasty (PTCA) to restore coronary flow
- Thrombolytic therapy to dissolve clots
2. Hypertension
2.1 Renal Artery Stenosis and Two-Kidney Hypertension
When one renal artery is stenotic but the contralateral kidney is normal:
- The ischemic kidney secretes excess renin → angiotensin II → aldosterone
- The contralateral (normal) kidney is exposed to high arterial pressure from angiotensin II
- It responds by pressure natriuresis, attempting to normalize BP
- A state of elevated renin-dependent hypertension results
- Known as renovascular hypertension or Goldblatt hypertension
PART II: RESPIRATORY SYSTEM PATHOLOGY
3. Respiratory Insufficiency - Principles of Diagnosis
Respiratory diseases result from:
- Inadequate ventilation
- Abnormalities of diffusion across the pulmonary membrane
- Abnormal blood transport of gases
Key diagnostic parameters:
- Blood gas analysis (PO2, PCO2, pH)
- Blood pH measured by glass electrode (Henderson-Hasselbalch principle)
- Blood CO2 measured via bicarbonate equilibrium: pH = 6.1 + log (HCO3⁻/CO2)
- Blood PO2 measured by polarography (platinum electrode, current proportional to O2 concentration)
- Pulmonary function tests: vital capacity, tidal volume, FRC, dead space, physiological shunt
4. Chronic Pulmonary Emphysema
Definition: "Excess air in the lungs" - a complex obstructive and destructive process.
Etiology: Years of cigarette smoking; chronic exposure to air pollution, dust, or chemical fumes.
Pathogenesis (Sequential):
- Chronic infection - irritants in smoke cause:
- Partial paralysis, then destruction of respiratory cilia
- Excess mucus secretion
- Inhibition of alveolar macrophages → reduced infection defense
- Infection + mucus + inflammatory edema → chronic obstruction of small airways
- Obstruction causes air trapping (especially on expiration) → alveolar overdistension → destruction of up to 50-80% of alveolar walls
Physiological Effects:
- Increased airway resistance → greatly increased work of breathing; expiration especially difficult (bronchioles collapse during expiratory effort)
- Loss of alveolar walls → reduced diffusing capacity → impaired O2 uptake and CO2 removal
- Ventilation-perfusion (V/Q) mismatch:
- Low V/Q zones → physiological shunt → poor blood oxygenation
- High V/Q zones → physiological dead space → wasted ventilation
- Loss of alveolar walls = loss of pulmonary capillaries → pulmonary hypertension → right heart overload → cor pulmonale (right heart failure)
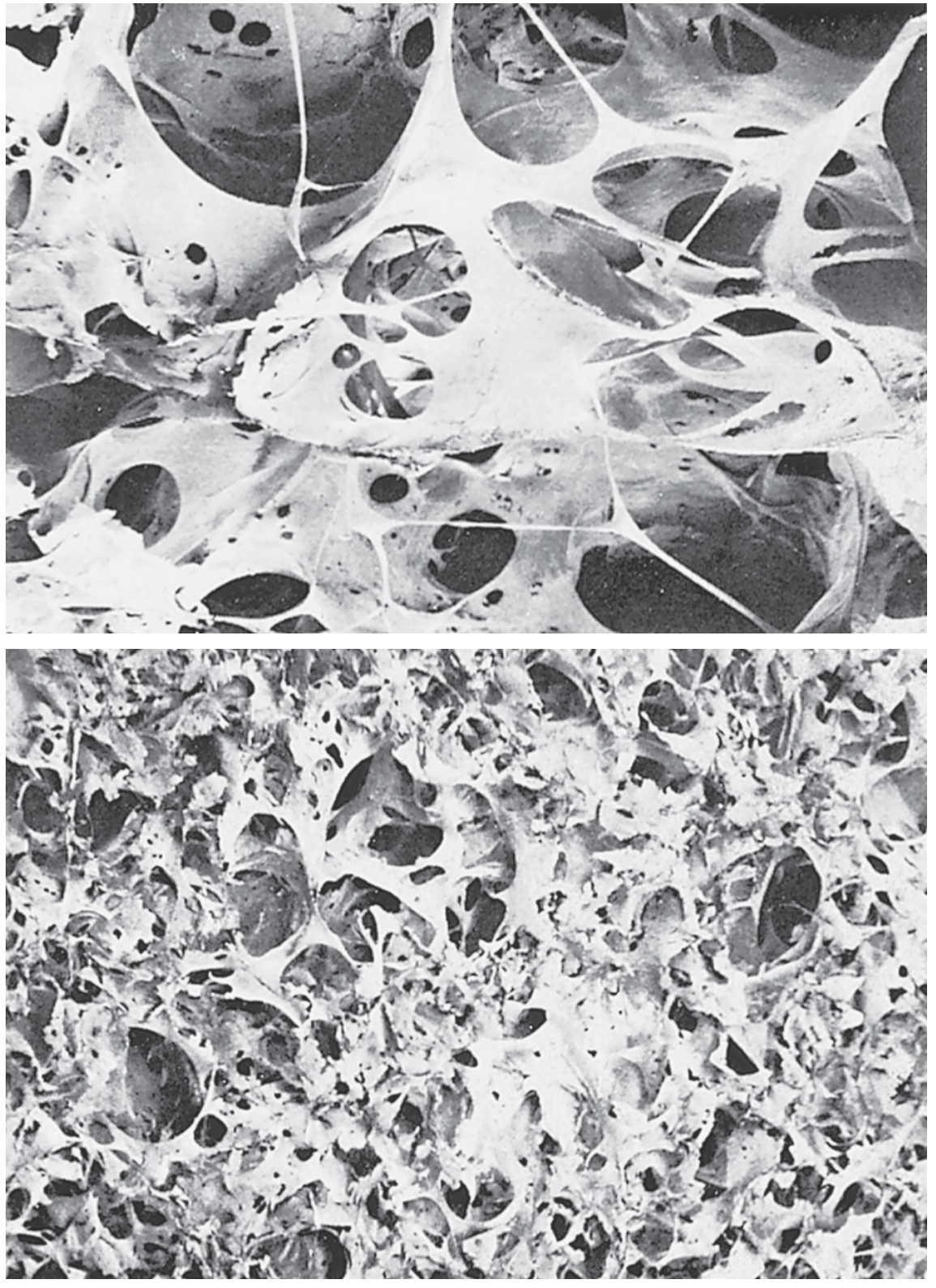
Figure: Contrast of emphysematous vs. normal lung tissue (Guyton & Hall, Fig. 43.4)
5. Bronchial Asthma
Prevalence: ~7-8% of US population; over 262 million affected worldwide (WHO estimate).
Pathophysiology:
- Contractile hypersensitivity of bronchioles to foreign substances
- ~70% of patients under 30: allergic (extrinsic) asthma - sensitivity to pollens, dust mites
- Older patients: hypersensitivity to non-allergenic irritants in smog (intrinsic/non-allergic asthma)
Mechanism (Allergic Asthma):
- Person forms excess IgE antibodies against specific allergens
- IgE attaches to mast cells in lung interstitium near bronchioles and small bronchi
- On allergen exposure, antigen-IgE reaction triggers mast cell degranulation, releasing:
- Histamine
- Slow-reacting substance of anaphylaxis (SRSA) - mixture of leukotrienes (LTC4, LTD4, LTE4)
- Eosinophilic chemotactic factor
- Bradykinin
- Combined effects → (a) localized edema in bronchiolar walls + thick mucus secretion; (b) bronchial smooth muscle spasm
Functional Consequences:
- Greatly increased airway resistance
- Expiration more affected than inspiration (external pressure collapses already-narrowed bronchioles)
- Markedly reduced maximum expiratory flow rate and reduced timed expiratory volume (FEV1)
- Dyspnea (air hunger)
- Increased FRC and residual volume (air trapping)
- Chronic: permanent "barrel chest" with chronically elevated FRC and residual volume
6. Tuberculosis
Pathology:
Mycobacterium tuberculosis causes:
- Tissue invasion by macrophages
- Granuloma formation (tubercle) - fibrous tissue walls off the lesion, limiting spread
In ~3% of untreated cases, walling-off fails → tubercle bacilli spread throughout both lungs → extreme tissue destruction with large abscess cavities.
Late-Stage Pulmonary Effects:
- Extensive fibrosis → increased work of breathing, reduced vital capacity and breathing capacity
- Reduced total respiratory membrane surface area → impaired gas diffusion
- V/Q mismatch from irregular fibrosis
- If severe: hypoxemia, pulmonary hypertension, right heart failure
7. Pulmonary Edema
Pathophysiology:
Fluid accumulates in pulmonary interstitium and alveoli when:
- Pulmonary capillary hydrostatic pressure exceeds plasma oncotic pressure (normal capillary pressure ~7 mm Hg)
- In left heart failure: pulmonary capillary pressure may rise to 30-40 mm Hg
- Once capillary pressure exceeds ~28-30 mm Hg, rapid flooding of alveoli occurs
Clinical result: Impaired gas exchange → hypoxemia → respiratory failure.
PART III: RENAL AND URINARY SYSTEM PATHOLOGY
8. Overview of Kidney Diseases
Chronic kidney disease (CKD) affects >14% of US adults (~35.5 million people). Kidney diseases are classified as:
- Acute Kidney Injury (AKI) - abrupt loss of function within days
- Chronic Kidney Disease (CKD) - progressive loss of nephron function
Sub-categories by location: vascular, glomerular, tubular, interstitial, obstructive (ureter/bladder).
9. Acute Kidney Injury (AKI)
9.1 Classification
| Category | Cause | Examples |
|---|---|---|
| Prerenal AKI | Decreased blood supply to kidneys (origin outside kidneys) | Heart failure, hemorrhage, severe dehydration |
| Intrarenal AKI | Abnormalities within the kidney | Glomerulonephritis, tubular necrosis, interstitial nephritis |
| Postrenal AKI | Obstruction of urinary collecting system | Kidney stones (calcium, urate, cystine), bladder outlet obstruction |
Note: In sepsis, prerenal and intrarenal causes often co-exist (low BP + endothelial/tubular injury simultaneously).
9.2 Prerenal AKI
- Kidneys normally receive 1100 mL/min (~20-25% of cardiac output)
- Reduced blood flow → reduced GFR → oliguria (urine output below fluid intake)
- Complete cessation of urine = anuria
- As long as renal blood flow does not fall below ~20-25% of normal, AKI can be reversed if cause is corrected before cell damage occurs
- Key protective mechanism: as GFR falls, less sodium chloride is filtered → less needs to be reabsorbed → less O2 consumed by tubules
- If blood flow falls below the basal metabolic requirement of tubular cells (usually <20-25% of normal), tubular cells become hypoxic and may die
- Prolonged ischemia → transition to intrarenal AKI (tubular necrosis)
9.3 Intrarenal AKI
Subdivisions:
- Conditions damaging glomerular capillaries/small vessels
- Conditions damaging renal tubular epithelium (acute tubular necrosis - ATN)
- Conditions damaging renal interstitium
9.4 AKI Caused by Acute Glomerulonephritis
- Most common type of intrarenal AKI caused by immune injury to glomeruli
- In ~95% of cases: immune-mediated damage occurs 1-3 weeks after infection elsewhere in the body (commonly streptococcal throat infection - "post-streptococcal GN")
- Mechanism: Antigen-antibody complexes deposit in glomerular membrane → activate complement → inflammatory cell infiltration → membrane thickening and damage
- Results in: protein in urine (proteinuria), red cells in urine (hematuria), reduced GFR, fluid retention, hypertension
Progression:
- Most patients recover (especially children)
- ~1% progress to rapidly progressive (crescentic) GN → chronic renal failure
9.5 Chronic Kidney Disease (CKD)
- Progressive nephron loss, with remaining nephrons hypertrophying to compensate
- CKD is classified in stages by GFR (Stage 1-5, where Stage 5 = GFR <15 mL/min = kidney failure requiring dialysis or transplant)
Consequences of severe CKD:
- Uremia (accumulation of urea, creatinine, other nitrogenous wastes)
- Fluid and electrolyte imbalances (hyperkalemia, metabolic acidosis, fluid overload)
- Renal anemia (reduced erythropoietin production)
- Renal osteodystrophy (impaired vitamin D activation → hypocalcemia → secondary hyperparathyroidism → bone disease)
- Hypertension (RAAS activation, fluid retention)
- Cardiovascular disease (accelerated atherosclerosis)
9.6 Glomerular Injury as a Cause of Chronic Renal Disease
- Repeated episodes of glomerular injury or progressive glomerulosclerosis → irreversible reduction in GFR
- The podocyte (visceral epithelial cell of the glomerular capillary) is key - podocyte loss = permanent glomerular scarring
- Even after the initial insult resolves, secondary hyperfiltration in remaining nephrons accelerates further damage
PART IV: GASTROINTESTINAL AND LIVER PATHOLOGY
10. Peptic Ulcer Disease
Definition: Excoriated area of stomach or intestinal mucosa caused by the digestive action of gastric juice.
Most frequent sites:
- Within a few centimeters of the pylorus (most common)
- Lesser curvature of the antral stomach
- Lower esophagus (acid reflux zone)
- Marginal ulcers after gastrojejunostomy
Normal Protective Mechanisms:
- Mucous barrier - mucus from gastric glands, mucous neck cells, pyloric glands, Brunner glands of duodenum
- Alkaline duodenal secretions:
- Pancreatic sodium bicarbonate neutralizes HCl and inactivates pepsin
- Bile and Brunner gland secretions
- Feedback control:
- Acid in duodenum inhibits gastric secretion and peristalsis (neural + hormonal)
- Acid releases secretin → pancreatic bicarbonate secretion (additional neutralization)
Pathogenesis: Peptic ulcer results from an imbalance between:
- Acid/pepsin secretion (aggressive factors), AND
- Mucosal protective mechanisms (defensive factors)
Specific Causes:
1. Helicobacter pylori infection:
- Gram-negative bacterium colonizes the mucous layer of gastric mucosa
- Produces urease → splits urea into NH3 + CO2 → locally destroys the mucous barrier
- Also produces cytotoxins directly damaging mucosal cells
- H. pylori infection is found in ~70-85% of gastric ulcer patients and ~95% of duodenal ulcer patients
- Eradication with antibiotics heals ulcers and dramatically reduces recurrence
2. Excess secretion of gastric acid and pepsin:
- Zollinger-Ellison syndrome: gastrin-secreting tumors (gastrinomas) in pancreas or duodenum → massively elevated acid secretion → multiple, refractory peptic ulcers
3. NSAID use:
- NSAIDs inhibit cyclooxygenase (COX-1) → reduced prostaglandin synthesis
- Prostaglandins normally maintain mucosal blood flow, stimulate mucus and bicarbonate secretion, and inhibit acid secretion
- Their reduction impairs mucosal defenses
11. Gastrointestinal Obstruction
Cause: Can occur at any point along the GI tract, from the esophagus to the rectum.
Pathophysiology:
- Obstruction of the pylorus or small intestine leads to projectile vomiting and progressive distension
- Intestinal obstruction → bacterial overgrowth → translocation of bacteria → sepsis
- Proximal dilation + increased intraluminal pressure → ischemia → necrosis → perforation
Consequences vary by location:
- Pyloric obstruction: metabolic alkalosis (loss of HCl in vomitus), hypokalemia
- Small intestinal obstruction: mixed metabolic disturbances, severe fluid and electrolyte loss
12. Malabsorption Syndromes
12.1 Celiac Disease (Non-Tropical Sprue)
- Autoimmune-mediated enteropathy triggered by gluten (protein in wheat, rye, barley)
- More common in women
- Mechanism: gluten elicits autoimmune reaction → inflammation → destruction of intestinal enterocytes
- Mild form: only microvilli destroyed → 2-fold reduction in absorptive surface area
- Severe form: villi completely blunted or absent → severe malabsorption
Consequences of severe celiac disease:
- Steatorrhea (fat malabsorption - fat appears in stools as fatty acid salts)
- Protein-energy malnutrition with severe bodily wasting
- Osteomalacia - bone demineralization (calcium malabsorption)
- Impaired coagulation (vitamin K malabsorption)
- Macrocytic anemia - pernicious type (B12 and folic acid malabsorption)
Treatment: Remove wheat and rye from diet - leads to recovery within weeks, especially in children.
12.2 Tropical Sprue
- Occurs in tropical regions
- Believed to be caused by intestinal bacterial infections causing mucosal inflammation
- Treated with antibacterial agents
13. Large Intestine Disorders
13.1 Constipation
- Slow movement of feces through the large intestine
- Causes: excessive fluid absorption from feces, insufficient fluid intake, obstruction (tumors, adhesions, ulcers), voluntary suppression of defecation reflexes
- Chronic inhibition of defecation reflexes eventually leads to loss of reflex sensitivity
13.2 Diarrhea
- Rapid movement of feces
- Causes: intestinal infection, inflammation, osmotic agents, increased motility
- Risks: dehydration, electrolyte loss (hypokalemia, metabolic acidosis)
SUMMARY TABLE - Key Pathological Mechanisms
| System | Condition | Core Mechanism |
|---|---|---|
| Cardiovascular | Acute Heart Failure | Sudden pump failure → sympathetic activation → compensation |
| Cardiovascular | Cardiogenic Shock | Cardiac output insufficient for survival; vicious cycle of hypotension-ischemia |
| Cardiovascular | HFpEF | Diastolic dysfunction; stiff LV, elevated filling pressures |
| Cardiovascular | Cardiomegaly | Chronic volume/pressure overload → myocardial stretching + fibrosis |
| Respiratory | Emphysema | Alveolar wall destruction → reduced diffusion, air trapping, cor pulmonale |
| Respiratory | Asthma | IgE-mediated mast cell degranulation → bronchospasm + mucosal edema |
| Respiratory | Tuberculosis | Granuloma formation; late fibrosis → restrictive pattern + V/Q mismatch |
| Renal | Prerenal AKI | Reduced renal perfusion → oliguria/anuria; reversible if corrected early |
| Renal | Glomerulonephritis | Immune complex deposition → glomerular inflammation and damage |
| Renal | CKD | Progressive nephron loss → uremia, electrolyte disturbance, renal anemia |
| GI | Peptic Ulcer | H. pylori / excess acid vs. mucosal defense imbalance |
| GI | Celiac Disease | Autoimmune gluten-mediated villous atrophy → malabsorption |
| GI | GI Obstruction | Mechanical blockage → distension, ischemia, potential necrosis |
Source: Guyton and Hall Textbook of Medical Physiology (9780443111013), Chapters 22, 24, 32, 43, 67-68
These notes cover all four systems you requested at a full textbook depth. Key highlights:
- Cardiovascular: Heart failure (acute, compensated, decompensated, HFpEF, high-output), cardiogenic shock with treatment, renovascular hypertension
- Respiratory: Emphysema pathogenesis and physiology, asthma (IgE/mast cell mechanism), tuberculosis, pulmonary edema
- Renal: AKI classification (pre/intra/post-renal), glomerulonephritis mechanism, CKD consequences
- GI: Peptic ulcer (H. pylori, NSAIDs, Zollinger-Ellison), celiac disease, GI obstruction, diarrhea/constipation
give me notes from robins text as of now for systemic pathology include every detail and stains used
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Here are your full, detailed notes from Robbins, Cotran & Kumar - Pathologic Basis of Disease (latest edition), covering all four major systems with every morphological detail, stain, mechanism, and clinical feature:
SYSTEMIC PATHOLOGY - ROBBINS & KUMAR
(Pathologic Basis of Disease, Latest Edition)
PART I: CARDIOVASCULAR PATHOLOGY
1. ISCHEMIC HEART DISEASE (IHD)
Definition: Group of related entities from myocardial ischemia - an imbalance between myocardial oxygen supply (perfusion) and demand. Ischemia is worse than hypoxemia alone because it also deprives the myocardium of nutrients and prevents removal of metabolic wastes.
Epidemiology: Single largest cause of mortality worldwide - >15% of global deaths. In high-resource countries ~9 million deaths/year. US death rate has fallen >50% since the 1960s due to risk factor modification, statins, thrombolysis, angioplasty/stenting, CABG, and better heart failure therapies.
Etiology:
-
90% due to obstructive atherosclerotic lesions in epicardial coronary arteries (CAD)
- Other causes: coronary emboli, vasculitis, vascular spasm, severe demand states (hypertrophy, tachycardia, shock)
- Note: Tachycardia is doubly harmful - increases O2 demand AND reduces coronary perfusion time (diastole shortened)
Clinical Presentations:
- Myocardial infarction (MI) - ischemic necrosis
- Angina pectoris - ischemia without infarction
- Chronic IHD with heart failure
- Sudden cardiac death (SCD)
1A. Myocardial Infarction (MI)
Types:
- STEMI (transmural) - complete occlusion, usually plaque rupture + thrombosis
- NSTEMI/UA (subendocardial) - incomplete occlusion, or demand > supply
Pathogenesis:
- Chronic atherosclerotic plaque builds up over decades
- Plaque disruption (rupture or erosion) exposes subintimal collagen/lipid core
- Platelet aggregation + thrombus formation → acute occlusion
- Ischemia → necrosis within 20-40 minutes if flow not restored
Morphology of MI - TIME-DEPENDENT CHANGES:
| Time | Gross | Microscopy | Key Stains |
|---|---|---|---|
| 0-30 min | Normal | Normal (wavy fibers) | Electron microscopy shows mitochondrial swelling |
| 1-3 hr | Normal | Waviness of fibers at border | H&E |
| 4-12 hr | Dark mottling (occasional) | Early coagulation necrosis; edema; hemorrhage; contraction bands | H&E - eosinophilic cytoplasm, nuclear changes (pyknosis) |
| 12-24 hr | Dark mottling | Ongoing coagulation necrosis; pyknosis; early neutrophil infiltration | H&E |
| 1-3 days | Tan/yellow center; hyperemic border | Heavy neutrophil infiltration; nuclear debris; cytolysis | H&E (neutrophils prominent) |
| 3-7 days | Hyperemic border; central yellow-tan softening | Macrophages appear; dead neutrophils; phagocytosis begins | H&E |
| 1-2 weeks | Yellow-tan; depressed, soft | Granulation tissue at margins; macrophages, capillaries, fibroblasts | H&E |
| Weeks-months | Gray-white scar | Progressive fibrosis; scarring complete | Masson Trichrome (collagen = blue) |
Special Stains for early MI detection (autopsy <12 hrs):
- Triphenyltetrazolium chloride (TTC) - infarcted tissue fails to take up stain (remains pale/white), viable tissue stains brick-red
- Nitroblue tetrazolium (NBT) - similar principle; dead tissue pale
Complications of MI:
- Arrhythmias (most common cause of death in first 24 hrs)
- Cardiogenic shock
- Cardiac rupture - free wall (days 3-7; hemopericardium, tamponade), interventricular septum (VSD), papillary muscle (mitral regurgitation)
- Ventricular aneurysm (late complication)
- Fibrinous pericarditis (Dressler syndrome - weeks later, immune mediated)
- Mural thrombus → systemic embolism
- Heart failure
2. VALVULAR HEART DISEASE
Types:
- Stenosis - failure to open completely; virtually always chronic (fibrosis/calcification)
- Insufficiency (regurgitation) - failure to close; can be acute or chronic
Most Frequent Acquired Valvular Lesions:
| Lesion | Most Common Cause |
|---|---|
| Aortic stenosis | Calcification of normal or bicuspid aortic valve |
| Aortic insufficiency | Dilation of ascending aorta (hypertension, aging) |
| Mitral stenosis | Rheumatic heart disease |
| Mitral insufficiency | Myxomatous degeneration (MVP) or LV dilation |
Hemodynamic consequences:
- Stenosis → pressure overload → concentric hypertrophy
- Insufficiency → volume overload → eccentric hypertrophy → both eventually → heart failure
2A. Calcific Aortic Stenosis
Morphology:
- Heaped-up calcified masses on outflow surface of aortic cusps
- Cusps remain mobile but calcification protrudes into sinuses, preventing opening
- Stain: Von Kossa stain (calcium deposits appear black), H&E (calcification appears as dark blue/basophilic)
2B. Rheumatic Heart Disease (Mitral Stenosis)
Pathogenesis: Molecular mimicry - Group A streptococcal M protein cross-reacts with cardiac proteins → autoimmune damage
Acute Rheumatic Fever - Morphology:
- Aschoff bodies - pathognomonic; foci of fibrinoid necrosis with lymphocytes, plasma cells, and Aschoff cells (plump macrophages with "owl-eye" nuclei and "caterpillar" chromatin)
- Anitschkow cells (caterpillar cells) - activated macrophages, pathognomonic for rheumatic carditis
- Pancarditis: pericarditis, myocarditis, endocarditis
- McCallum plaques on posterior left atrial wall
- Fibrinous pericarditis ("bread-and-butter" pericarditis)
- Vegetations: small, warty vegetations along line of valve closure
- Stain: H&E shows Aschoff bodies well
Chronic Rheumatic Heart Disease - Morphology:
- Leaflet thickening and retraction
- Commissural fusion (classic "fish-mouth" or "buttonhole" orifice of mitral valve)
- Chordal thickening and fusion
- Stain: H&E shows dense fibrosis and calcification
3. CARDIOMYOPATHY
Definition: Intrinsic cardiac muscle diseases - genetic or from well-defined causes. Three pathophysiologic categories:
3A. Dilated Cardiomyopathy (DCM) - 90% of cardiomyopathies
- Pathophysiology: Systolic (contractile) dysfunction
- Causes:
- Up to 50%: autosomal dominant loss-of-function mutations in cytoskeletal/sarcolemmal/nuclear envelope genes; titin (TTN) truncations account for ~20%
- Myocarditis (viral - especially coxsackievirus B)
- Toxic (alcohol, doxorubicin/anthracyclines, cocaine)
- Peripartum cardiomyopathy
- Hemochromatosis, thiamine deficiency
- Morphology:
- Massive 4-chamber dilation; weight >900 g (normal ~300 g)
- Thin ventricular walls
- Mural thrombi common (especially in LV apex)
- Histology: myocyte hypertrophy, increased interstitial fibrosis, only scattered myocyte necrosis
- Stains: H&E (myocyte changes), Masson Trichrome (fibrosis = blue)
3B. Hypertrophic Cardiomyopathy (HCM)
- Pathophysiology: Diastolic (relaxation) dysfunction
- Genetics: Autosomal dominant gain-of-function mutations in sarcomeric proteins; most common: beta-myosin heavy chain and myosin-binding protein C
- Morphology:
- Massive cardiac hypertrophy (often asymmetric - especially septum: ASH - asymmetric septal hypertrophy)
- Banana-shaped LV cavity
- Histology - PATHOGNOMONIC: myocyte hypertrophy + myofiber disarray (chaotic, haphazard arrangement of myocytes and myofibrils within cells) + interstitial fibrosis
- Stain: H&E shows myofiber disarray; Masson Trichrome shows fibrosis
3C. Restrictive Cardiomyopathy
- Pathophysiology: Stiff, noncompliant myocardium; diastolic dysfunction
- Causes:
- Amyloid deposition - most common
- Radiation-induced fibrosis
- Endomyocardial fibrosis (tropical; eosinophilia)
- Sarcoidosis
- Hemochromatosis
- Amyloid (Cardiac):
- Stain: Congo Red - salmon-pink on H&E, apple-green birefringence under polarized light (pathognomonic)
- Grossly: stiff, rubbery myocardium; "waxy" appearance
3D. Myocarditis
- Causes: Viral (coxsackievirus B, HIV, parvovirus B19), bacterial, parasitic (T. cruzi - Chagas), autoimmune
- Morphology:
- Active myocarditis (Dallas criteria): interstitial lymphocytic inflammatory infiltrate + adjacent myocyte necrosis/damage
- Giant cell myocarditis: multinucleated giant cells with lymphocytes, eosinophils - poor prognosis
- Stain: H&E is key; immunohistochemistry for T-cell markers
4. PERICARDIAL DISEASE
- Pericardial Effusion: Normal <50 mL; >200-300 mL rapid accumulation → cardiac tamponade
- Hemopericardium: Blood in pericardium; causes: MI wall rupture, aortic dissection
- Fibrinous Pericarditis:
- Causes: uremia, MI (Dressler syndrome), rheumatic fever
- Gross: "bread-and-butter" pericarditis (shaggy fibrinous exudate)
- Stain: H&E shows fibrin; PTAH (phosphotungstic acid-hematoxylin) stains fibrin blue
- Constrictive Pericarditis: Fibrous obliteration of pericardial sac → cardiac compression; causes: TB, pyogenic
PART II: RESPIRATORY PATHOLOGY
5. EMPHYSEMA
Definition: Irreversible enlargement of airspaces distal to the terminal bronchiole, with destruction of alveolar walls (without fibrosis).
Types (Anatomic Classification):
| Type | Location of Destruction | Main Cause | Notes |
|---|---|---|---|
| Centriacinar (centrilobular) | Proximal acinus (respiratory bronchioles); distal alveoli spared | Cigarette smoking (>95% of cases) | Upper lobe predominant; apical segments |
| Panacinar (panlobular) | Entire acinus uniformly | Alpha-1 antitrypsin (AAT) deficiency; also smoking | Lower lobe predominant; bases |
| Paraseptal (distal acinar) | Distal acinus; near pleura/septa | Cause of spontaneous pneumothorax in young adults | - |
| Irregular | Around scars | Post-inflammatory scarring | Not clinically significant |
Pathogenesis - "Protease-Antiprotease Hypothesis":
- Smoking/irritants → chronic inflammation → neutrophils/macrophages release elastase, metalloproteinases
- Normally countered by alpha-1 antitrypsin (AAT) and other antiproteases
- Protease-antiprotease imbalance → elastin breakdown in alveolar walls → destruction
- In AAT deficiency: genetic; severe, early-onset panacinar emphysema; AAT gene = SERPINA1 (chromosome 14)
- Smoking also causes oxidative stress directly damaging antiproteases
Morphology:
- Gross: Voluminous, pale lungs; do not collapse; bullae (large air spaces) especially in paraseptal type; barrel chest on CXR
- Microscopy:
- Enlarged airspaces with thin, delicate walls
- Loss of alveolar septa
- Reduced capillary bed
- No significant fibrosis (distinguishes from fibrosis)
- Stains: H&E (loss of septa seen); Elastic-van Gieson (EVG) - highlights elastin loss in alveolar walls (elastin = black/dark purple); Verhoeff-van Gieson (VVG)
Clinical: Dyspnea (predominant), minimal cough, "pink puffer" (pursed-lip breathing, barrel chest, hyperinflated, maintain PaO2), cor pulmonale in late stage.
6. CHRONIC BRONCHITIS
Definition (Clinical): Persistent productive cough for at least 3 months in at least 2 consecutive years.
Pathogenesis: Smoking → chronic mucus hypersecretion → Reid index increase (mucous gland thickness: total bronchial wall thickness >50%)
Morphology:
- Gross: Hyperemia, edema, mucosal secretions; thickened bronchial wall
- Microscopy:
- Hypertrophy and hyperplasia of mucous glands (submucosal) in trachea/bronchi
- Goblet cell metaplasia in small airways (normally no goblet cells there)
- Reid index = (mucous gland thickness)/(total bronchial wall thickness) - normally <0.4; in chronic bronchitis >0.5
- Inflammatory infiltrate (lymphocytes, macrophages); neutrophils in acute exacerbations
- Squamous metaplasia of bronchial epithelium
- Stains: H&E for Reid index measurement; Alcian blue/PAS for mucus
7. BRONCHIAL ASTHMA
Definition: Chronic inflammatory disorder of the airways characterized by recurrent, reversible bronchospasm.
Types:
- Atopic (extrinsic/allergic) asthma - most common; IgE-mediated; type I hypersensitivity; begins in childhood
- Non-atopic (intrinsic) asthma - non-immune; triggered by respiratory infections, irritants, cold, exercise
- Drug-induced asthma - especially aspirin (aspirin-sensitive asthma); blocks COX → leukotrienes > prostaglandins; no IgE involvement
- Occupational asthma - fumes, organic/chemical dusts
Pathogenesis (Atopic):
- Prior sensitization → production of IgE antibodies against allergen
- IgE binds to mast cells in bronchial submucosa
- Re-exposure: allergen cross-links IgE → mast cell degranulation
- Early phase (minutes): histamine, prostaglandins, leukotrienes (LTC4, LTD4, LTE4) → bronchoconstriction, mucus secretion, vasodilation
- Late phase (4-8 hrs): eosinophil/neutrophil recruitment → eosinophilic inflammation → epithelial damage; major basic protein (MBP) from eosinophils → further injury
Morphology:
- Gross: Lungs overinflated; airways plugged with thick, viscid mucus
- Microscopy:
- Curschmann spirals - mucous plugs with epithelial cells (shed in sputum)
- Charcot-Leyden crystals - eosinophil membrane protein (elongated, double-pointed) in sputum
- Subepithelial fibrosis (airway remodeling)
- Basement membrane thickening ("subepithelial collagen deposition")
- Goblet cell hyperplasia
- Smooth muscle hypertrophy/hyperplasia
- Eosinophilic infiltrate (hallmark)
- Mucous gland hyperplasia
- Edema and vascular congestion
- Stains: H&E (eosinophils - bright pink granules; mast cells); Congo Red or Luna stain for eosinophils; PAS for mucus; Alcian blue for acidic mucins
8. PNEUMONIA
8A. Acute (Bacterial) Lobar Pneumonia
Most common organism: Streptococcus pneumoniae (lobar distribution)
Stages of lobar pneumonia:
- Congestion (1-2 days): congested, heavy lung; serous exudate; numerous bacteria; few neutrophils - Stain: H&E shows vascular congestion
- Red hepatization (2-4 days): lung red, firm, airless (like liver); alveoli filled with neutrophils, red cells, fibrin - Stain: H&E (pink fibrinous exudate with neutrophils)
- Gray hepatization (4-8 days): gray-brown color; RBCs lysed; fibrinopurulent exudate persists; macrophages appear - Stain: H&E
- Resolution (8+ days): exudate enzymatically digested; macrophages clear debris; normal architecture restored
Gram stain: Gram-positive diplococci (pneumococcus)
8B. Bronchopneumonia (Lobular Pneumonia)
- Patchy consolidation of multiple lobules
- Associated with H. influenzae, S. aureus, K. pneumoniae, P. aeruginosa
- Histology: neutrophilic infiltration centered on bronchioles/bronchi, spreading to alveoli
Key histologic distinction:
- Bacterial pneumonia: intraalveolar neutrophilic inflammation
- Viral pneumonia: interstitial lymphocytic inflammation
8C. Atypical (Interstitial) Pneumonia
- "Walking pneumonia" - mild symptoms despite radiographic infiltrates
- Causes: Mycoplasma pneumoniae, Chlamydophila pneumoniae, Legionella, viruses (influenza, COVID-19)
- Histology: interstitial inflammation (lymphocytes, macrophages); alveolar walls thickened; no intraalveolar exudate
- Legionella: silver stain (Warthin-Starry or Dieterle stain) to visualize organisms
8D. Aspiration Pneumonia
- Debilitated patients; gastric content aspiration
- Chemical + bacterial (mixed aerobic/anaerobic oral flora)
- Necrotizing pneumonia → lung abscess
- Microaspiration: non-necrotizing granulomas with multinucleated foreign body giant cells
8E. Lung Abscess
- Local suppurative necrosis of lung tissue
- Most common cause: aspiration (~60% are anaerobic organisms - Bacteroides, Fusobacterium, Peptococcus)
- Morphology: cavity filled with pus, fibrous wall
- Stains: H&E (necrosis + neutrophils); culture of organism required
9. INTERSTITIAL LUNG DISEASES
9A. Pulmonary Fibrosis (UIP/IPF)
- Usual Interstitial Pneumonia (UIP) is the histologic pattern of Idiopathic Pulmonary Fibrosis (IPF)
- Morphology:
- Temporal heterogeneity (old and new fibrosis coexist) - pathognomonic
- Dense subpleural fibrosis
- Honeycombing (cystic spaces lined by bronchiolar epithelium)
- Fibroblastic foci (active fibrosis - key diagnostic feature)
- Relatively mild inflammation
- Stains: H&E; Masson Trichrome (fibrosis = blue/green); EVG for elastic fibers
9B. Sarcoidosis
- Non-caseating granulomas involving lungs (and other organs)
- Morphology:
- Non-caseating granulomas: epithelioid macrophages + Langhans giant cells + lymphocytes; NO necrosis
- Schaumann bodies - laminated concentric calcified inclusions inside giant cells
- Asteroid bodies - stellate inclusions inside giant cells
- Stains: H&E (granulomas); ZN (Ziehl-Neelsen) or Fite used to exclude TB (negative in sarcoidosis); PAS or Grocott to exclude fungi
9C. Malignant Mesothelioma
- Aggressive tumor of pleural mesothelium; linked to asbestos exposure (latency 25-45 years)
- Morphology:
- Grows as sheets/nodules encasing lung; tumor "rind" on pleura
- Three histologic subtypes: epithelioid (most common; best prognosis), sarcomatoid, biphasic
- Epithelioid: tubular/papillary structures resembling adenocarcinoma
- Key stains to distinguish from adenocarcinoma:
- Positive in mesothelioma: calretinin (nuclear + cytoplasmic), WT-1, CK5/6, D2-40 (podoplanin), mesothelin
- Negative in mesothelioma (positive in adenocarcinoma): CEA, TTF-1, CD15, MOC-31, BerEP4
- PASD (PAS with diastase) - neutral mucin positive in adenocarcinoma; negative in mesothelioma
PART III: RENAL / URINARY PATHOLOGY
10. GLOMERULAR DISEASES
Structure of Glomerulus:
- Glomerular filtration barrier: fenestrated endothelium + GBM (collagen IV, laminin, heparan sulfate) + podocyte foot processes
- Charge barrier (anionic heparan sulfate in GBM) prevents albumin passage
- Mesangial cells: phagocytic, structural, contractile
Clinical Syndromes:
| Syndrome | Features |
|---|---|
| Nephritic syndrome | Hematuria, RBC casts, proteinuria <3.5 g/day, hypertension, oliguria, azotemia |
| Nephrotic syndrome | Proteinuria >3.5 g/day, hypoalbuminemia, edema, hyperlipidemia, lipiduria |
| Rapidly progressive GN | Acute nephritis + rapid loss of renal function (weeks to months) |
| Chronic GN | End-stage glomerular disease |
| Asymptomatic hematuria/proteinuria | IgA nephropathy, thin basement membrane disease |
10A. Acute Proliferative (Post-Streptococcal) Glomerulonephritis
Cause: Group A beta-hemolytic Streptococcus (nephritogenic strains M types 1, 2, 4, 12)
Pathogenesis: Immune complex deposition (type III hypersensitivity) → complement activation → neutrophil infiltration
Morphology:
- Light microscopy: Enlarged, hypercellular glomeruli (diffuse proliferative); hypercellularity due to: (1) infiltrating leukocytes (neutrophils + monocytes); (2) proliferation of endothelial and mesangial cells; (3) in severe cases, crescents
- Immunofluorescence (IF): Granular ("lumpy-bumpy") deposits of IgG and C3 along GBM and mesangium - "starry sky" pattern
- Electron microscopy (EM): Subepithelial electron-dense "humps" (immune complex deposits)
Stains: H&E (hypercellularity); Periodic Acid-Schiff (PAS) (highlights GBM); Masson Trichrome (immune deposits appear red/fuchsinophilic); Jones Methenamine Silver (JMS) (GBM black, immune deposits appear as holes)
Clinical:
- Children: hematuria ("smoky/cola urine"), periorbital edema, hypertension, oliguria; 1-2 weeks post-sore throat
- Labs: elevated ASO titer, low C3, RBC casts in urine
-
95% children recover; adults have worse prognosis
10B. Rapidly Progressive (Crescentic) Glomerulonephritis (RPGN)
Definition: Nephritic syndrome + rapid deterioration of renal function (days to weeks); crescents in >50% of glomeruli
Three Types:
| Type | Mechanism | IF Pattern | Example |
|---|---|---|---|
| Type I (Anti-GBM) | Linear IgG against GBM (collagen IV alpha-3 chain) | Linear IgG along GBM | Goodpasture syndrome (lung + kidney) |
| Type II (Immune complex) | Granular IF deposits | Granular deposits | Post-streptococcal GN, lupus, IgA |
| Type III (Pauci-immune) | No IF deposits; ANCA-mediated | No deposits | GPA (c-ANCA/PR3), MPA (p-ANCA/MPO) |
Morphology:
- Crescents = proliferating parietal epithelial cells + infiltrating monocytes/macrophages filling Bowman space
- Fibrin deposition within crescents
- Stain: H&E (crescents); PAS (Bowman capsule highlighted); Fibrin stain (PTAH or MSB) shows fibrin in crescents
10C. Nephrotic Syndrome - Major Causes
Pathophysiology of Nephrotic Syndrome:
- Deranged glomerular permeability → protein leakage
- Massive proteinuria (>3.5 g/day) → hypoalbuminemia → decreased oncotic pressure → edema + sodium retention
- Compensatory hepatic lipoprotein synthesis → hyperlipidemia → lipiduria
- Loss of immunoglobulins → susceptibility to infections (especially pneumococcus, staphylococcus)
- Loss of antithrombin III + protein C/S → hypercoagulability → renal vein thrombosis, DVT, PE
Urinary findings:
- Oval fat bodies (lipoproteins reabsorbed by tubular cells) - Maltese cross pattern under polarized light
- Fatty casts
- Stain: Oil Red O or Sudan IV for fat in urine/tissue
10D. Minimal Change Disease (MCD) / Lipoid Nephrosis
Most common cause of nephrotic syndrome in children (<8 years)
Pathogenesis:
- Dysfunction of T-cells → circulating "permeability factor" → podocyte injury
- Loss of slit diaphragm proteins (nephrin, podocin)
- Selective proteinuria (mainly albumin loss)
- Associated with atopy, Hodgkin lymphoma (paraneoplastic)
Morphology:
- Light microscopy: NORMAL glomeruli (hence "minimal change")
- IF: NEGATIVE (no immune deposits)
- EM: DIFFUSE EFFACEMENT (fusion) of podocyte foot processes - pathognomonic
- Stains: H&E (normal LM); EM is essential for diagnosis
- Excellent response to corticosteroids
10E. Focal Segmental Glomerulosclerosis (FSGS)
Most common cause of nephrotic syndrome in adults (especially Black Americans)
Types:
- Primary (idiopathic) - podocyte injury
- Secondary: HIV (HIVAN), heroin, sickle cell, obesity, congenital nephron loss
Morphology:
- Light microscopy: Focal (some glomeruli) and segmental (part of each glomerulus) sclerosis - collapse of capillary loops with increased matrix; hyalinosis; foam cells
- Perihilar variant (most common), tip variant (best prognosis), collapsing variant (worst prognosis; HIVAN)
- IF: Nonspecific IgM and C3 in sclerotic segments
- EM: Diffuse foot process effacement (like MCD) + focal basement membrane thickening
- Stains: PAS (sclerosis highlighted by PAS-positive matrix increase); JMS/PASM (Jones methenamine silver) (silver stains GBM black; sclerotic areas show collapse)
10F. Membranous Nephropathy
Most common cause of nephrotic syndrome in adults >50 (especially white males)
Pathogenesis:
- Most cases (~70-80%): anti-phospholipase A2 receptor (PLA2R) antibodies → subepithelial immune complex deposits
- Secondary: SLE, hepatitis B, drugs (penicillamine, gold), malignancy
Morphology:
- LM: Diffuse thickening of GBM; "spikes" projecting from GBM around subepithelial deposits
- IF: Granular IgG (and C3) along GBM in a subepithelial pattern (characteristic)
- EM: Subepithelial electron-dense deposits; GBM spikes between deposits
- Stain: JMS (Jones Silver) - GBM black; deposits appear as "spikes" and "holes" (holes = where deposits were, spikes = new GBM around deposits); PAS highlights thickened GBM
Clinical: Heavy proteinuria; rule of thirds - 1/3 spontaneous remission, 1/3 persistent proteinuria but stable, 1/3 progress to renal failure
10G. Membranoproliferative GN (MPGN)
Pathogenesis:
- Type I: Immune complex mediated (subendothelial deposits; Hep B, Hep C, cryoglobulinemia)
- Type II (Dense Deposit Disease - DDD): Complement dysregulation; C3 nephritic factor
Morphology:
- LM: Mesangial cell proliferation + GBM thickening; "lobular" appearance; "tram-track" or "double-contour" GBM (pathognomonic) - due to mesangial interposition between endothelium and GBM
- IF: Granular C3 ± IgG (Type I); C3 alone (Type II/DDD)
- EM: Type I - subendothelial deposits; Type II - dense deposits within GBM (ribbon-like)
- Stain: PAS and JMS - tram-tracking of GBM highlighted; Masson Trichrome - deposits appear fuchsinophilic
10H. IgA Nephropathy (Berger Disease)
Most common form of GN worldwide
Pathogenesis:
- Aberrant glycosylation of IgA1 (deficient O-linked galactosylation in hinge region)
- Aberrantly glycosylated IgA is deposited in mesangium
- Alternative complement pathway activation (C3 present; C1q and C4 absent)
- Associated with celiac disease, liver disease, HSP (systemic form)
Morphology:
- LM: Mesangial proliferation and increased matrix; mesangioproliferative pattern
- IF: IgA (dominant), often with IgG, IgM, C3 in mesangium - pathognomonic
- EM: Electron-dense mesangial deposits
- Stains: H&E (mesangial expansion); PAS (matrix increase); IF is diagnostic
Clinical: Episodic hematuria 1-2 days after upper respiratory infections (synpharyngitic hematuria) - classic
10I. Lupus Nephritis
WHO/ISN Classification (1-VI):
- Class III (focal proliferative) and Class IV (diffuse proliferative) - most severe
- Class V (membranous) - nephrotic syndrome
Morphology (Class IV):
- Diffuse endocapillary and mesangial proliferation
- "Wire-loop" lesions: subendothelial deposits create thickened GBM
- IF: "Full house" pattern - IgG, IgA, IgM, C3, C1q all positive (characteristic of lupus)
- EM: Subendothelial deposits (wire loops), mesangial deposits, "fingerprint" deposits (tubuloreticular inclusions) in endothelial cells (due to IFN-alpha)
- Stains: H&E, PAS, JMS, Trichrome - all used; IF critical
11. TUBULAR AND INTERSTITIAL DISEASES
11A. Acute Tubular Injury (ATI) / Acute Tubular Necrosis (ATN)
Causes:
- Ischemic ATN: prolonged hypoperfusion (shock, sepsis, severe hemorrhage)
- Nephrotoxic ATN: aminoglycosides, contrast agents, cisplatin, heavy metals, myoglobin (rhabdomyolysis), hemoglobin
Morphology:
- Ischemic ATN: focal tubular necrosis with skip lesions; most severe at corticomedullary junction (where straight proximal tubule and thick ascending limb are hypoxia-sensitive); tubular rupture (tubulorrhexis); casts in distal tubules; vascular congestion
- Nephrotoxic ATN: uniform proximal tubular damage; intact tubular basement membrane (no tubulorrhexis)
- Stains: H&E (tubular cell loss, nuclear necrosis); PAS (loss of brush border in proximal tubules)
PART IV: GASTROINTESTINAL PATHOLOGY
12. ESOPHAGUS
12A. Barrett Esophagus
Definition: Intestinal metaplasia of esophageal squamous mucosa - complication of chronic GERD; premalignant
Morphology:
- Gross: Red, velvety "tongues" extending upward from GEJ; alternates with pale squamous mucosa
- Microscopy:
- Intestinal-type metaplasia: goblet cells with mucus vacuoles that stain pale blue (conspicuous among gastric-type foveolar cells)
- Goblet cells are diagnostic - classic wine-goblet shape with pale cytoplasm
- Low-grade vs. high-grade dysplasia: nuclear hyperchromasia, increased N:C ratio, failure of maturation
- High-grade dysplasia: atypical mitoses, gland budding/irregularity, cellular crowding
- Stains: H&E (goblet cells identified by their pale blue mucin); Alcian blue (pH 2.5) - goblet cell mucin = bright blue (intestinal-type mucin); PAS - foveolar cell mucin = magenta
13. STOMACH
13A. Gastritis
Acute Gastritis/Gastropathy:
- NSAIDs: inhibit COX-1 → ↓ prostaglandins (PGE2, PGI2) → ↓ mucus, bicarbonate, mucosal blood flow → mucosal erosion
- Alcohol, bile, stress: disrupt tight junctions + increase acid back-diffusion
- Morphology (H&E): erosion (loss of surface epithelium), neutrophils in lamina propria, vascular congestion
Chronic Gastritis (Type A vs. Type B):
| Feature | Type A (Autoimmune) | Type B (H. pylori) |
|---|---|---|
| Location | Fundus/body | Antrum |
| Cause | Anti-parietal cell antibodies; anti-intrinsic factor | H. pylori infection |
| Association | Pernicious anemia, other autoimmune diseases | Peptic ulcer, gastric cancer, MALT lymphoma |
| Parietal cells | Lost | Relatively preserved |
H. pylori Gastritis - Morphology:
- H&E: Dense lymphoplasmacytic infiltrate in lamina propria + neutrophils in epithelium and gland lumina ("active" chronic gastritis); lymphoid follicle formation
- Warthin-Starry stain or Giemsa stain - demonstrates H. pylori organisms (curved gram-negative rods) in mucous layer
- Modified Giemsa stain - most commonly used in clinical practice for H. pylori
- Urease test (CLO test): rapid bedside test (H. pylori produces urease → converts urea to ammonia → pH rise → color change)
14. SMALL INTESTINE AND COLON
14A. Inflammatory Bowel Disease (IBD)
Two major types:
| Feature | Crohn Disease (CD) | Ulcerative Colitis (UC) |
|---|---|---|
| Location | Anywhere from mouth to anus; ileum most common | Colon only; rectum always involved; contiguous |
| Distribution | Skip lesions (transmural) | Continuous (mucosal/submucosal) |
| Depth | Transmural inflammation | Mucosal + submucosal |
| Ulcers | Linear, "cobblestone" | Broad, superficial; pseudopolyps |
| Wall | Thickened, rubbery "hose-pipe" | Thin; dilated (toxic megacolon) |
| Granulomas | Yes - NON-CASEATING granulomas (~50%) | NO granulomas |
| Fistulae/Strictures | Common | Rare |
| Cancer risk | Slightly increased | Greatly increased (colitis-associated dysplasia) |
| Serology | Anti-Saccharomyces cerevisiae antibodies (ASCA) | p-ANCA |
Crohn Disease - Morphology:
- Transmural inflammation with all wall layers involved
- Non-caseating granulomas (50%; even in normal-looking areas outside bowel)
- Linear, deep "knife-like" ulcers
- Cobblestone mucosa (islands of intact mucosa surrounded by ulcers)
- Fissures and fistulae
- Fat wrapping (creeping fat over serosal surface)
- Stains: H&E (granulomas - epithelioid macrophages + Langhans giant cells); ZN stain to exclude TB (negative in CD)
UC - Morphology:
- Mucosal and submucosal inflammation only
- Crypt abscesses (neutrophils filling crypts) - hallmark
- Cryptitis (neutrophils invading crypt epithelium)
- Goblet cell depletion (mucus depletion)
- Pseudopolyps (islands of regenerating mucosa)
- No granulomas
- Paneth cell metaplasia in left colon
- Stains: H&E (crypt abscesses + goblet cell depletion); PAS/Alcian blue for mucus depletion; Movat pentachrome for wall layers
15. LIVER
15A. Cirrhosis
Definition: Diffuse hepatic fibrosis with parenchymal nodular regeneration. End-stage liver disease.
Causes (worldwide):
- Chronic viral hepatitis (B and C) - worldwide leading cause
- Alcohol-associated liver disease
- Metabolic dysfunction-associated steatotic liver disease (MASLD/MAFLD - formerly NASH)
- Autoimmune hepatitis
- Primary biliary cholangitis (PBC)
- Primary sclerosing cholangitis (PSC)
- Metabolic (hemochromatosis, Wilson disease, AAT deficiency)
Morphology:
- Gross: Bumpy, nodular liver surface; nodules separated by fibrous bands; late stage: shrunken liver
- Microscopy:
- Parenchymal nodules (regenerative; lack central veins) surrounded by dense fibrous bands
- Two types: micronodular (<3 mm - alcohol-related) and macronodular (>3 mm - viral hepatitis)
- Mixed pattern in advanced disease
- Stains:
- H&E - baseline; shows hepatocyte changes
- Masson Trichrome - fibrosis/collagen = blue (key stain for staging fibrosis)
- Sirius Red - fibrosis red (quantitative; used in research)
- Reticulin (Gordon-Sweet silver) - normal lobular architecture highlighted; loss of reticulin in nodules shows regeneration
- PAS with diastase (PASD) - Mallory-Denk bodies in alcohol-related cirrhosis; PAS-positive diastase-resistant granules in AAT deficiency (stored AAT globules in hepatocytes)
- Prussian Blue (Perls stain) - iron = blue (hemochromatosis)
- Rhodanine or Rubeanic acid stain - copper = orange-red (Wilson disease)
- Orcein or Victoria Blue stain - HBsAg-containing hepatocytes; AAT globules
Consequences of Cirrhosis:
- Portal hypertension → esophageal varices, splenomegaly, caput medusae, ascites
- Hepatic failure → coagulopathy, jaundice, encephalopathy, hypoalbuminemia
- Hepatorenal syndrome
- Increased risk of hepatocellular carcinoma (HCC) - especially Hep B + C
15B. Viral Hepatitis
Hepatitis A & E: Acute only; fecal-oral; never chronic
Hepatitis B, C, D: Can cause chronic hepatitis → cirrhosis → HCC
Acute Hepatitis Morphology:
- Hepatocyte ballooning degeneration
- Acidophil (Councilman/Mallory) bodies - pyknotic shrunken hepatocytes
- Lobular disarray with necrosis
- Kupffer cell hyperplasia
- Portal and lobular inflammation (lymphocytes)
- Stains: H&E (ballooning, necrosis, inflammation); Immunohistochemistry (IHC) for HBsAg, HBcAg (chronic HBV)
Chronic Hepatitis:
- Portal and periportal inflammation (lymphocytes, plasma cells)
- Interface hepatitis (piecemeal necrosis of hepatocytes at portal-parenchymal interface) - marker of activity
- Bridging necrosis (portal to portal or portal to central necrosis) - severe disease
- Stains: H&E; Masson Trichrome for fibrosis staging; Orcein/Victoria Blue for ground-glass hepatocytes (HBsAg)
15C. Alcoholic Liver Disease
Spectrum:
- Hepatic steatosis (fatty liver) - reversible; >5% hepatocytes contain fat
- Alcoholic hepatitis - acute; can be fatal
- Alcoholic cirrhosis - irreversible end-stage
Morphology of Alcoholic Hepatitis:
- Macrovesicular steatosis (large fat vacuoles displacing nucleus to periphery)
- Mallory-Denk bodies (Mallory hyaline) - tangled intermediate filaments (cytokeratin 8/18) in hepatocyte cytoplasm; eosinophilic, irregular, rope-like inclusions - key finding
- Neutrophilic (not lymphocytic) lobular inflammation
- Hepatocyte ballooning degeneration
- Pericellular/perisinusoidal fibrosis ("chicken wire" fibrosis)
- Stains: H&E (Mallory-Denk bodies = eosinophilic); Masson Trichrome (pericellular fibrosis = blue); Ubiquitin immunostain (Mallory-Denk bodies = positive)
PART V: SPECIAL STAINS MASTER TABLE
| Stain | What it Shows | Color | Used In |
|---|---|---|---|
| H&E | Routine tissue; nuclei/cytoplasm | Blue/pink | All diagnoses |
| PAS (Periodic Acid-Schiff) | Glycogen, glycoproteins, basement membranes, fungi, mucins | Magenta/purple | GBM in GN, mucus, fungi (Aspergillus, Mucor), glycogen storage |
| PASD (PAS + diastase) | Same as PAS but glycogen removed; AAT globules remain | Magenta | AAT deficiency (PAS+ diastase-resistant) |
| Masson Trichrome | Collagen/fibrosis | Blue/green | Fibrosis staging (liver, heart, kidney) |
| Sirius Red | Collagen | Red | Fibrosis (quantitative) |
| Jones Methenamine Silver (JMS/PASM) | GBM (black); highlights holes/spikes | Black GBM | Membranous nephropathy, MPGN, FSGS |
| Congo Red | Amyloid | Salmon-pink (H&E); apple-green birefringence under polarized light | Amyloidosis (all types) |
| Prussian Blue (Perls) | Iron/hemosiderin | Blue | Hemochromatosis, hemosiderosis |
| Rhodanine / Rubeanic acid | Copper | Orange-red | Wilson disease |
| Orcein / Victoria Blue | HBsAg; elastic fibers; AAT | Brown/dark | Chronic HBV, AAT deficiency, vasculature |
| Reticulin (Gordon-Sweet) | Reticular fibers/Type III collagen | Black | Liver architecture, lymphoma, tumors |
| Masson-Fontana | Melanin; argentaffin granules | Black | Melanoma, carcinoid tumors |
| Ziehl-Neelsen (ZN/AFB) | Acid-fast bacteria | Red on blue | TB, MAC infection |
| Giemsa / Modified Giemsa | H. pylori; mast cells; parasites | Blue/purple | H. pylori gastritis, malaria |
| Warthin-Starry | Spirochetes; H. pylori; Legionella | Black | H. pylori, Bartonella, syphilis |
| Gram stain | Gram+/- bacteria | Blue/red | Pneumonia organisms, endocarditis |
| Grocott Methenamine Silver (GMS) | Fungi | Black | Aspergillus, Candida, Mucor, PJP |
| Alcian blue | Acid mucins (pH 2.5) | Bright blue | Barrett goblet cells; Crohn mucin |
| Oil Red O / Sudan IV | Neutral fats/lipids | Red | Fatty liver, fatty embolism, oval fat bodies |
| Von Kossa | Calcium (phosphate) | Black | Calcification (aortic stenosis, metastatic calcification) |
| Alizarin Red | Calcium (carbonate) | Red-orange | Calcification |
| Elastic-Van Gieson (EVG) / Verhoeff-van Gieson (VVG) | Elastic fibers | Black | Emphysema, vasculature, aneurysm |
| Toluidine Blue / Luna stain | Mast cells; eosinophils | Metachromatic (purple-red for mast cells) | Asthma, carcinoid, mastocytosis |
| Immunofluorescence (IF) | IgG, IgA, IgM, C3, C1q (glomeruli) | Green fluorescence | All GN classification |
| Ubiquitin IHC | Mallory-Denk bodies; Lewy bodies | Brown | Alcoholic hepatitis |
| PTAH (phosphotungstic acid-hematoxylin) | Fibrin; striations | Blue | Fibrin in crescents, pericarditis |
Source: Robbins, Cotran & Kumar Pathologic Basis of Disease (ISBN 9780443264528), Chapters 12, 15, 17, 18, 20
These notes cover the full depth of Robbins systemic pathology with:
- Every major disease and its pathogenesis
- Gross and microscopic morphology for each condition
- Every special stain with colors and diagnostic context
- Clinical features and syndrome correlations
- Immunofluorescence patterns for all major glomerular diseases
- Detailed cardiomyopathy genetics and histology
- Complete GI spectrum from Barrett esophagus to IBD to cirrhosis
Search for high-resolution microscopy images of key pathologic findings (emphysema, glomerulonephritis, cirrhosis) and also include chapters 8 14 15 16 17 18 19 20 21 22 notes
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Loading Image
Loading Image
Loading Image
Loading Image
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Reading File
ROBBINS & KUMAR - PATHOLOGIC BASIS OF DISEASE
Chapters 8, 14–22 | Systemic Pathology with Stains & Microscopy Images
CHAPTER 8 - NEOPLASIA
Key Concepts: Benign vs. Malignant Tumors
| Feature | Benign | Malignant |
|---|---|---|
| Differentiation | Well differentiated; resembles tissue of origin | Poorly differentiated to anaplastic |
| Function | Often retains normal function | May acquire unexpected functions |
| Growth rate | Slow | Generally faster |
| Border | Circumscribed; usually encapsulated | Poorly circumscribed; invasive |
| Spread | Remains localized | Metastasizes to distant sites |
| Route of spread | - | Carcinomas → lymphatics; Sarcomas → hematogenous |
Epidemiology of Cancer (2023 US Data)
- Estimated cancer deaths worldwide (2020): 9.9 million (~1 in 6 deaths)
- Projected to reach 13.2 million by 2030
- Males: prostate, lung, colon/rectum
- Females: breast, lung, colon/rectum
- These four cancers = >50% of US diagnoses and deaths
Molecular Basis of Cancer
Key genetic alterations:
- Proto-oncogene mutations → gain-of-function (RAS, MYC, HER2)
- Tumor suppressor gene loss → loss-of-function (TP53, RB1, APC, BRCA1/2)
- DNA repair gene mutations → microsatellite instability (MLH1, MSH2 - Lynch syndrome)
- Telomerase activation → immortality
- Epigenetic alterations → methylation silencing of tumor suppressors
Hallmarks of Cancer (Hanahan & Weinberg):
- Self-sufficiency in growth signals
- Insensitivity to growth inhibitors
- Evasion of apoptosis
- Limitless replicative potential
- Sustained angiogenesis
- Tissue invasion and metastasis
- Reprogramming of energy metabolism
- Evading immune destruction
Grading and Staging
- Grade: Degree of differentiation (I - well differentiated to IV - anaplastic)
- Stage: Extent of spread (TNM: Tumor size, Node status, Metastasis)
- Staging is more clinically important for prognosis
CHAPTER 14 - BLOOD VESSELS
Atherosclerosis
Definition: Intimal-based lesion of large/medium arteries composed of a fibrous cap + atheromatous core (lipid, necrotic debris, foam cells, calcification, inflammatory cells, SMCs).
Epidemiology: Causes ~half of all deaths in Western countries. #1 worldwide cause of MI, stroke, peripheral vascular disease.
Risk Factors:
| Nonmodifiable | Modifiable |
|---|---|
| Genetic variation / family history | Hyperlipidemia (LDL most important) |
| Increasing age | Hypertension |
| Male sex | Smoking |
| Diabetes/insulin resistance | |
| Obesity | |
| Physical inactivity | |
| Elevated CRP (inflammation) |
Pathogenesis - "Response to Injury" Hypothesis:
- Endothelial injury/dysfunction (from hyperlipidemia, HTN, smoking, toxins)
- Endothelial dysfunction → increased permeability + VCAM-1/ICAM-1 expression → monocyte recruitment
- Monocytes enter intima → become macrophages → ingest oxidized LDL → foam cells
- Smooth muscle cells (SMCs) migrate from media to intima, proliferate, produce ECM → fibrous cap
- Lipid core accumulates with foam cell death, cholesterol crystals, calcification
- Plaque grows → luminal stenosis OR plaque rupture → thrombosis
Plaque Types:
- Stable plaque: dense fibrous cap, minimal lipid, little inflammation - produces chronic ischemia
- Vulnerable/unstable plaque: thin cap, large lipid core, dense macrophage infiltrate - prone to rupture → acute MI/stroke
Morphology:
- Gross: fatty streaks (earliest lesion, reversible) → raised yellow-white plaques → ulcerated/calcified plaques
- LM: intimal thickening, foam cells (lipid-laden macrophages), fibrous cap (collagen, SMCs), necrotic core
- Stains: H&E (foam cells); Masson Trichrome (fibrous cap = blue collagen); Oil Red O/Sudan on frozen sections (lipid = red); Von Kossa (calcification = black); EVG (elastic lamina disruption)
Aneurysms
Definition: Localized abnormal dilation of a blood vessel or heart.
- True aneurysm: involves all layers of intact (but thinned) wall (atherosclerotic, congenital, post-MI ventricular)
- False aneurysm (pseudoaneurysm): wall defect → extravascular hematoma communicating with lumen (post-MI rupture contained by pericardium; graft anastomosis leak)
- Dissection: blood enters a wall defect and tunnels through medial planes; can occur without aneurysm
Types by shape: saccular (spherical outpouching, part of wall) vs. fusiform (symmetric dilation, whole circumference)
Abdominal Aortic Aneurysm (AAA):
- Most >55 years; M>F; strongly linked to smoking and atherosclerosis
-
90% below renal arteries
- Risk of rupture increases with size (>5.5 cm = surgical repair threshold)
- Morphology: atherosclerosis with thinned, focally destroyed media; aneurysm lined by mural thrombus
- Stain: EVG - elastic fiber fragmentation in media
Thoracic Aortic Aneurysm / Aortic Dissection:
- Causes: hypertension, Marfan syndrome (fibrillin-1 mutation), Ehlers-Danlos, bicuspid aortic valve
- Cystic medial degeneration - loss of elastic tissue and SMCs in aortic media (medial degeneration)
- Stain: EVG or Movat pentachrome - elastic fiber fragmentation + mucoid/cystic spaces in media
Vasculitis
Classification by vessel size:
Large vessel vasculitis:
-
Giant cell (temporal) arteritis:
- Chronic granulomatous inflammation of large-to-medium arteries
- Affects temporal, ophthalmic, vertebral arteries; almost always in >50 yr
- Granulomas with Langhans giant cells; involvement of internal elastic lamina
- Risk: blindness from ophthalmic artery involvement
- Stain: H&E (granulomas + giant cells); EVG (fragmented internal elastic lamina)
-
Takayasu arteritis:
- Granulomatous vasculitis affecting aorta and its main branches
- Young Asian women (<40); "pulseless disease"
- Irregular thickening of aortic wall with intimal hyperplasia; narrowing of branch ostia
- Stain: H&E (granulomas, giant cells); EVG (elastic fiber disruption)
Medium vessel vasculitis:
- Polyarteritis nodosa (PAN):
- Necrotizing vasculitis of medium-sized muscular arteries; spares pulmonary vasculature
- Associated with Hep B in ~30%
- Nodular thickening at branch points; aneurysmal dilation; NOT associated with ANCA
- Stain: H&E (fibrinoid necrosis of vessel wall, acute and chronic inflammation); Elastic stain (disruption of elastica)
Small vessel vasculitis (ANCA-associated):
-
Granulomatosis with Polyangiitis (GPA, formerly Wegener):
- Triad: upper respiratory necrotizing granulomas + lower respiratory granulomatous vasculitis + glomerulonephritis
- c-ANCA (PR3-ANCA) positive ~90%
- Saddle-nose deformity (cartilage destruction)
-
Microscopic Polyangiitis (MPA):
- Necrotizing vasculitis without granulomas; p-ANCA (MPO-ANCA) positive
- Pulmonary-renal syndrome (like GPA but no granulomas)
-
Eosinophilic Granulomatosis with Polyangiitis (EGPA/Churg-Strauss):
- Asthma + eosinophilia + granulomatous vasculitis
- p-ANCA positive in ~40%
CHAPTER 15 - THE HEART (see previous detailed notes from Chapter 12)
Additional: Congenital Heart Disease
Incidence (excluding trivial defects): ~1% of live births
Most common defects (frequency):
| Defect | % |
|---|---|
| Ventricular septal defect (VSD) | 42% |
| Atrial septal defect (ASD) | 10% |
| Pulmonary stenosis | 8% |
| Patent ductus arteriosus (PDA) | 7% |
| Tetralogy of Fallot | 5% |
| Coarctation of aorta | 5% |
Left-to-right shunts (acyanotic): VSD, ASD, PDA
- Initially acyanotic; over time: pulmonary hypertension → reversal → Eisenmenger syndrome (right-to-left shunt → cyanosis)
Right-to-left shunts (cyanotic at birth):
- Tetralogy of Fallot (most common cyanotic CHD): VSD + overriding aorta + pulmonary stenosis + RVH
- Transposition of great arteries
Obstructive lesions: Coarctation of aorta (male predominant; associated with Turner syndrome, bicuspid aortic valve)
CHAPTER 16 - RED CELLS, BLEEDING DISORDERS, LYMPHOID & MYELOID NEOPLASMS
Lymphoid Neoplasms
Classification (WHO):
- Precursor B-cell neoplasms (immature B cells)
- Peripheral B-cell neoplasms (mature B cells)
- Precursor T-cell neoplasms
- Peripheral T-cell/NK-cell neoplasms
- Hodgkin lymphoma
Leukemias vs. Lymphomas:
- Leukemias: widespread bone marrow + peripheral blood involvement
- Lymphomas: discrete tissue masses (usually lymph nodes)
- These are a continuum - many "lymphomas" can evolve to "leukemic" presentation
Key Lymphoid Neoplasms:
Hodgkin Lymphoma (HL):
- Pathognomonic cell: Reed-Sternberg (RS) cell - large, bilobed or multinucleated, "owl-eye" nucleoli; derived from germinal center B cells
- Stain: H&E (RS cells); CD30+ and CD15+ (immunohistochemistry); PAX5+ (weak B-cell marker)
- Four subtypes: nodular sclerosis (most common, young women, mediastinum) > mixed cellularity > lymphocyte-rich > lymphocyte-depleted
Diffuse Large B-Cell Lymphoma (DLBCL):
- Most common adult NHL (~30%)
- Large lymphoid cells with vesicular nuclei, multiple nucleoli
- CD20+, CD79a+; Germinal center B-cell type (GCB) vs. activated B-cell type (ABC)
- Stain: H&E (diffuse large cells); IHC panel: CD20, BCL-6, MUM-1, Ki-67
Follicular Lymphoma:
- Indolent; t(14;18)(q32;q21) - BCL-2/IgH translocation → BCL-2 overexpression → apoptosis block
- Morphology: follicular (nodular) pattern throughout lymph node; cells in follicles are centrocytes (small, cleaved) + centroblasts (large)
- Stain: H&E (follicular pattern); IHC: CD10+, BCL-6+, BCL-2+, CD20+
Burkitt Lymphoma:
- Highly aggressive; associated with EBV and HIV
- "Starry sky" pattern (macrophages ingesting apoptotic cells among sea of lymphoma cells)
- t(8;14) MYC/IgH translocation → MYC overexpression
- Stain: H&E (starry sky); IHC: CD20+, CD10+, BCL-6+, BCL-2-, Ki-67 ~100%
Multiple Myeloma:
- Plasma cell neoplasm; monoclonal Ig production (M spike on SPEP)
- "Fried-egg" or "clock-face" chromatin in malignant plasma cells
- Lytic bone lesions, hypercalcemia, renal failure (light chain cast nephropathy), anemia, infections
- Stain: H&E (plasma cells); IHC: CD138+, MUM-1+, CD38+; kappa or lambda light chain restriction by IHC or ISH
CHAPTER 17 - GASTROINTESTINAL TRACT
Esophagus
Barrett Esophagus (full detail in previous notes)
Goblet cells of Barrett: stain pale blue with H&E; bright blue with Alcian blue (pH 2.5) (intestinal-type acid mucin)
Stomach
Gastropathy/Gastritis:
- NSAIDs → inhibit COX-1 → ↓ PGE2, PGI2 → ↓ mucus, bicarbonate, mucosal blood flow → erosion
- Alcohol, bile, stress: disrupt tight junctions
- Morphology (H&E): erosion, mucosal congestion, neutrophilic infiltrate
H. pylori Gastritis:
- Curved gram-negative rods in mucous layer
- Dense lymphoplasmacytic lamina propria infiltrate + neutrophils in glands (active gastritis)
- Lymphoid follicle formation
- Stains: Modified Giemsa (organisms = blue, curved rods); Warthin-Starry (organisms = black); H&E (active chronic gastritis); CLO test (urease-based rapid test)
Peptic Ulcer Disease:
- Disrupted mucosal barrier → acid injury → ulcer
- Gross: sharply punched-out, smooth edges (benign), ~95% in duodenal bulb or gastric antrum
- Compare malignant ulcer: irregular heaped-up edges
- Histology (H&E): fibrinopurulent exudate → granulation tissue → fibrosis
Gastric Carcinoma:
- Two types:
- Intestinal type (Lauren): gland-forming, arises in intestinal metaplasia (H. pylori → chronic gastritis → metaplasia → dysplasia → carcinoma); hematogenous spread; better prognosis
- Diffuse type (Lauren): no gland formation; signet-ring cells (mucin pushes nucleus to periphery); linitis plastica (leather-bottle stomach); peritoneal dissemination; worse prognosis
- Stains: H&E (both types); PAS/Alcian blue (mucin in signet-ring cells); IHC: CK7, CK20, CDX2 (intestinal type)
Krukenberg Tumor: gastric carcinoma metastasizing to ovary (bilateral ovarian signet-ring cell deposits)
Small Intestine and Colon
Intestinal Obstruction - 4 main mechanical causes (Robbins Fig. 17.25):
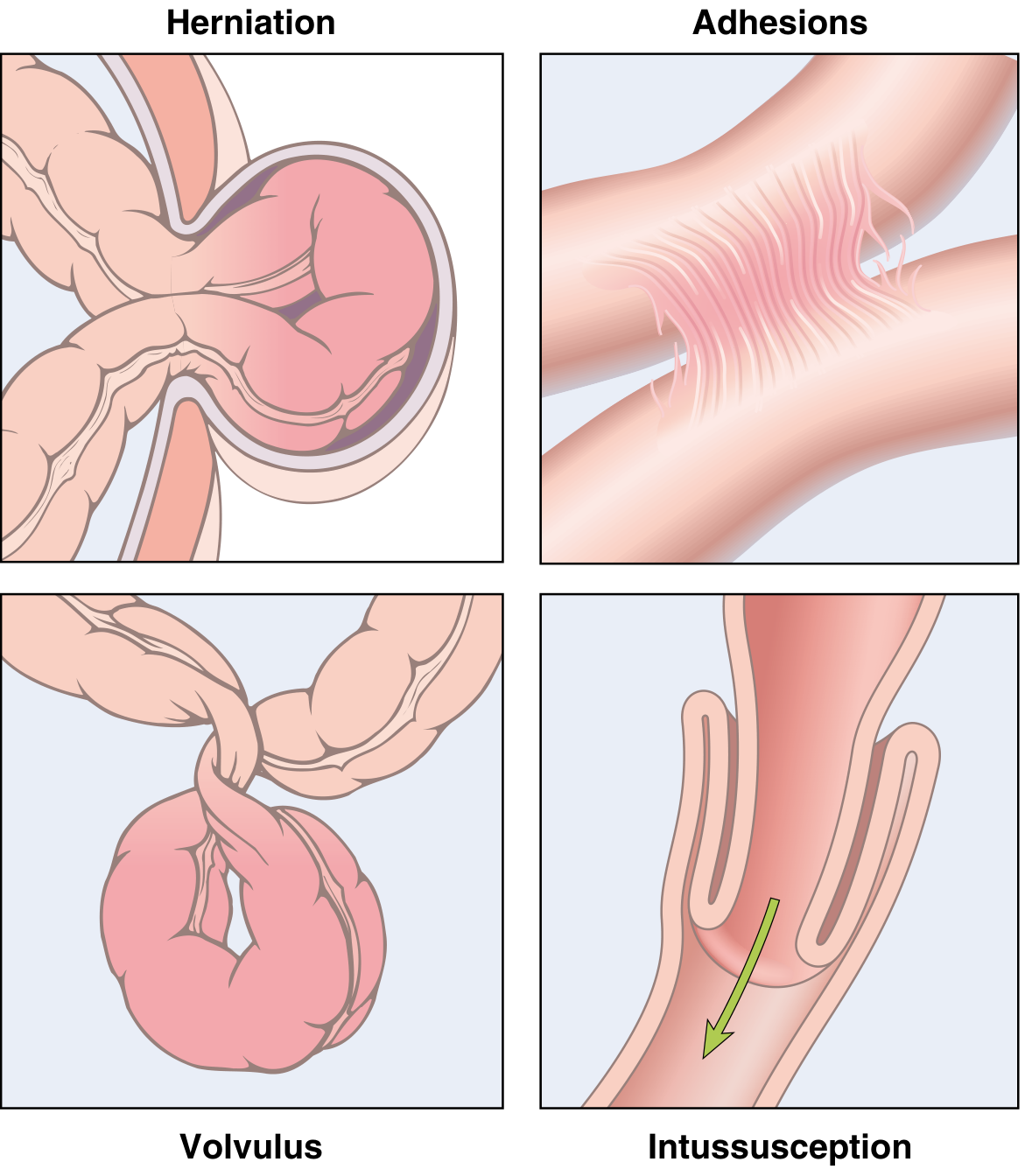
Fig. 17.25 (Robbins): (1) Hernia, (2) Adhesions, (3) Volvulus, (4) Intussusception
- Hernia - inguinal/femoral/umbilical (3% most common cause worldwide)
- Adhesions - post-surgical/inflammatory fibrous bridges
- Volvulus - loop twists on mesenteric axis; sigmoid or cecum most common
- Intussusception - telescoping; ileum into cecum most common; in children = lead point is Meckel's; adults = neoplasm
Inflammatory Bowel Disease: (detailed table in previous notes)
Colorectal Carcinoma:
- Most common GI malignancy in Western nations
- Two pathways:
- APC/beta-catenin pathway (80%): APC tumor suppressor loss → Wnt pathway activation → polyp → carcinoma (adenoma-carcinoma sequence, FAP)
- Microsatellite instability (MSI) pathway (15-20%): MLH1/MSH2 loss → Lynch syndrome or sporadic (MLH1 methylation)
- Gross: polypoid (right colon) vs. annular "napkin ring" constricting (left colon)
- Stains: H&E (gland-forming adenocarcinoma with necrotic luminal debris); IHC: CK20+, CDX2+, CK7- (colorectal pattern); MMR protein IHC (MLH1, MSH2, MSH6, PMS2 - loss of nuclear staining indicates MSI)
CHAPTER 18 - LIVER, GALLBLADDER, AND BILIARY TRACT
Cirrhosis
Morphology (Masson Trichrome - Fig. 18.7 from Robbins):
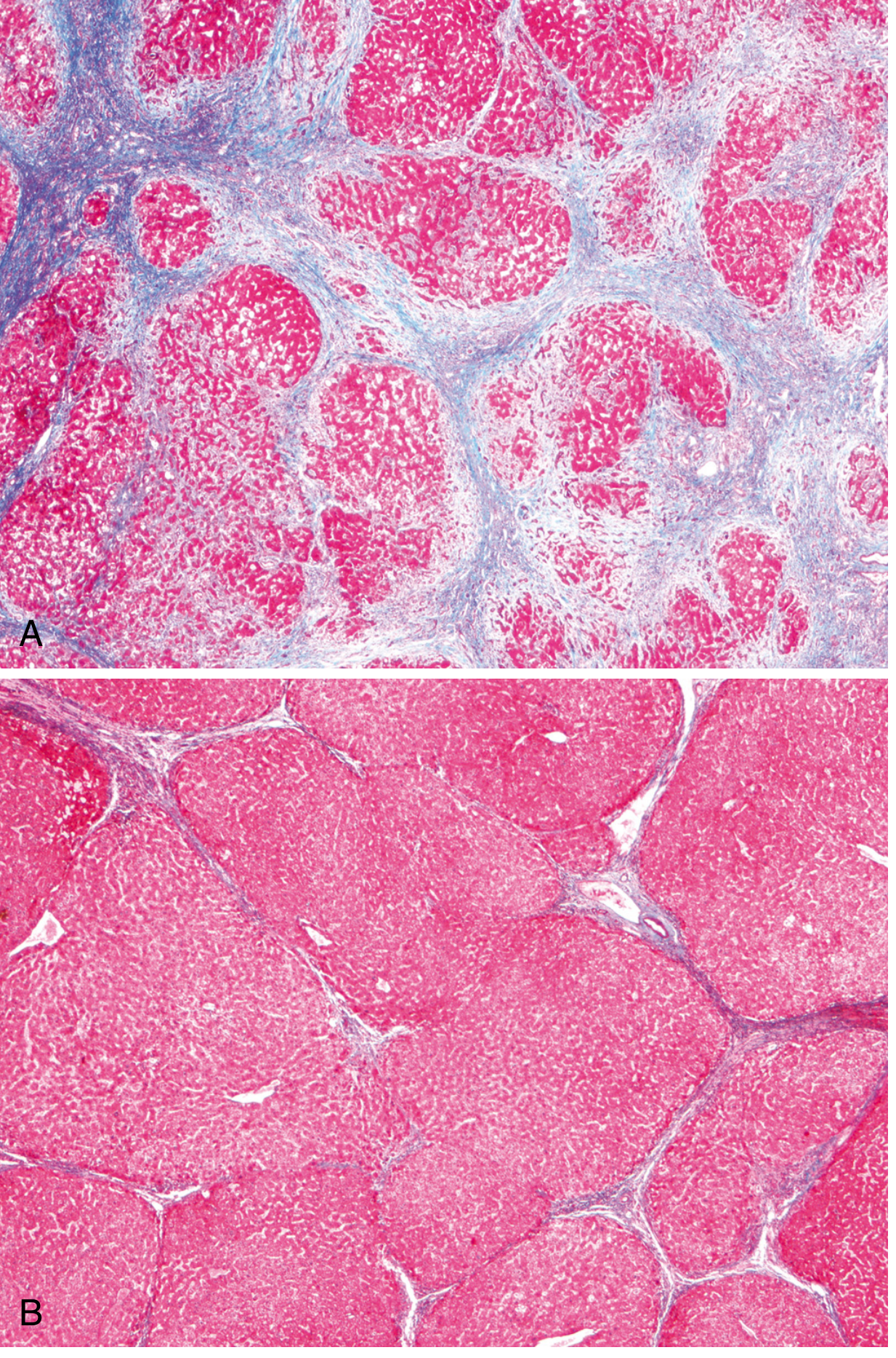
(A) Alcoholic cirrhosis: thick blue collagen bands surrounding red hepatocyte nodules. (B) After 1 year abstinence: thin incomplete scars showing regression. Masson Trichrome stain.
Clinical features of cirrhosis:
- 40% asymptomatic until advanced disease
- Cause of death: hepatic encephalopathy, variceal bleeding, infections, HCC
- Hyperestrogenemia → spider angiomas, palmar erythema, gynecomastia (males)
- Hypoalbuminemia → ascites, edema
- Pruritus from bile salt deposition
Alcoholic Liver Disease
Spectrum:
- Hepatic steatosis: >5% hepatocytes with fat; macrovesicular (large fat vacuole displacing nucleus); Oil Red O (fat = red); reversible
- Alcoholic hepatitis: macrovesicular steatosis + Mallory-Denk bodies + neutrophilic lobular inflammation + ballooning degeneration + satellite necrosis + pericellular "chicken wire" fibrosis
- Alcoholic cirrhosis: irreversible; micronodular initially; Masson Trichrome for fibrosis
Mallory-Denk Bodies:
- Tangled cytokeratin 8/18 intermediate filaments
- H&E: eosinophilic, rope-like cytoplasmic inclusions in balloned hepatocytes
- Ubiquitin IHC - positive (Mallory-Denk bodies are ubiquitinated)
- p62 IHC - also positive
Viral Hepatitis
| Virus | Transmission | Acute/Chronic | HCC risk | Serology |
|---|---|---|---|---|
| HAV | Fecal-oral | Acute only | No | Anti-HAV IgM (acute); IgG (past/immune) |
| HBV | Blood/sexual/perinatal | Both | High | HBsAg, HBeAg, HBcAg; anti-HBs (recovery/vaccine) |
| HCV | Blood | Both (chronic 80%) | High | Anti-HCV; HCV RNA |
| HDV | Co/superinfection with HBV | Both | - | Anti-HDV; requires HBV |
| HEV | Fecal-oral | Acute only (except immunocompromised) | No | Anti-HEV IgM |
Chronic HBV - "Ground-Glass Hepatocytes":
- Cytoplasm packed with smooth ER containing HBsAg
- H&E: finely granular, pale, ground-glass cytoplasm
- Orcein stain / Victoria Blue: HBsAg = brown
- IHC: HBsAg (cytoplasmic); HBcAg (nuclear or cytoplasmic in high replication)
Interface hepatitis (piecemeal necrosis):
- Periportal hepatocytes destroyed at interface with portal tract
- Indicates active disease and progressive fibrosis
Hepatocellular Carcinoma (HCC)
- Most common primary liver malignancy worldwide
- Strongly linked to: chronic HBV, chronic HCV, alcoholic cirrhosis, hemochromatosis, MASLD/NASH
- AFP (alpha-fetoprotein) elevated in ~75%
- Gross: single or multinodular; hemorrhagic/necrotic; portal vein invasion
- Morphology:
- Trabecular pattern most common (hepatocytes in cords separated by sinusoids)
- Pseudoglandular (acinar) pattern
- Scirrhous (fibrous stroma) pattern
- Well-differentiated HCC: polygonal cells with prominent nucleoli; retain hepatocytic features
- Stains: H&E (trabecular pattern); Reticulin (thickened trabeculae; normal liver = 1-2 cells thick, HCC = 3+ cells thick); IHC: HepPar-1, Arginase-1, GPC3 (glypican-3 - strong positive in HCC); CK7-, CK20-; AFP+
Primary Biliary Cholangitis (PBC)
- Autoimmune destruction of small intrahepatic bile ducts
- Middle-aged women; anti-mitochondrial antibodies (AMA) >95% sensitive/specific
- Morphology: florid duct lesion - lymphocytic infiltrate destroying bile duct epithelium; non-caseating granulomas around bile ducts
- Progression: chronic cholestasis → ductopenia → cirrhosis
- Stains: H&E (duct destruction); CK7 or CK19 IHC (highlights bile ducts)
Primary Sclerosing Cholangitis (PSC)
- Fibro-obliterative inflammation of intra- and extrahepatic bile ducts
- Strongly associated with ulcerative colitis (70-80%)
- p-ANCA positive
- "Onion-skin" periductal fibrosis - pathognomonic - concentric rings of fibrosis around bile ducts
- Stains: H&E + Masson Trichrome (periductal fibrosis = blue)
- High risk of cholangiocarcinoma
CHAPTER 19 - PANCREAS
Pancreatitis
Acute Pancreatitis - Etiology (GETSMASHED mnemonic):
- Gallstones (most common, ~40%)
- Ethanol/alcohol (second most common, ~35%)
- Trauma
- Steroids
- Mumps (and other viruses)
- Autoimmune (IgG4-related)
- Scorpion venom
- Hyperlipidemia / Hypercalcemia
- ERCP (iatrogenic)
- Drugs (azathioprine, statins, GLP-1 agonists)
Genetic associations:
- PRSS1 gain-of-function (cationic trypsinogen) - prevents trypsin self-inactivation → hereditary pancreatitis
- SPINK1 loss-of-function - trypsin inhibitor gone
- CFTR loss-of-function - inspissated secretions, duct obstruction
- CASR mutations - calcium dysregulation
Pathogenesis: Acinar cell injury → premature intracellular enzyme activation → autodigestion → fat necrosis + hemorrhage
Acute Pancreatitis Morphology:
- Interstitial (edematous) pancreatitis: edema, slight parenchymal inflammation, no necrosis; mild, self-limited
- Necrotizing pancreatitis (severe):
- Fat necrosis: chalky-white areas of saponification (calcium soaps from lipase digestion of mesenteric/peripancreatic fat)
- Hemorrhage: red-black hemorrhagic areas
- Microvascular injury → parenchymal necrosis
- Inflammatory infiltrate (initially neutrophilic)
- Stains: H&E (fat necrosis - ghost outlines of adipocytes, basophilic calcium deposits); Von Kossa (calcium = black in fat necrosis areas)
- Pseudocyst formation (late): cyst lined by fibrous wall, no epithelial lining
Chronic Pancreatitis:
- Recurrent or continuous inflammation → progressive fibrosis + destruction of acinar cells → exocrine insufficiency
- Causes: alcohol (most common in adults), hereditary, autoimmune, idiopathic
- Morphology: irregular fibrosis, acinar atrophy, ductal dilation and plugging with protein-rich secretions (calcifications in duct), relative preservation of islets
- Stains: H&E (fibrosis, acinar loss, preserved islets); Masson Trichrome (fibrosis = blue)
- Clinical: steatorrhea, malabsorption, diabetes (late - islet loss)
Pancreatic Carcinoma
- 4th leading cause of cancer death in US; 5-year survival <10%
-
90% = ductal adenocarcinoma
- Location: head of pancreas (60%) > body (15%) > tail (5%); 20% diffuse
- Precursor lesions: PanIN (Pancreatic Intraepithelial Neoplasia) 1→2→3 = carcinoma in situ; also IPMN, MCN
Molecular alterations (in order of progression):
- KRAS mutation (>90%; earliest and most common; codon 12)
- p16/CDKN2A loss (>90%; telomere shortening, cell cycle dysregulation)
- SMAD4 (DPC4) loss (~55%; TGF-beta pathway; loss = poor prognosis marker)
- TP53 loss (~75%; late event)
Morphology:
- Gross: hard, gray-white scirrhous mass; head lesions obstruct CBD → obstructive jaundice (Courvoisier sign)
- LM: moderately differentiated gland-forming adenocarcinoma in dense desmoplastic stroma; perineural invasion (characteristic)
- Stains: H&E (glands in desmoplastic stroma); IHC: CK7+, CK20-, CA19-9+, CEA+; SMAD4 loss by IHC (absent nuclear staining)
CHAPTER 20 - KIDNEY AND URINARY TRACT
Emphysema - Microscopy Images from Robbins:
Diagram of emphysema types:
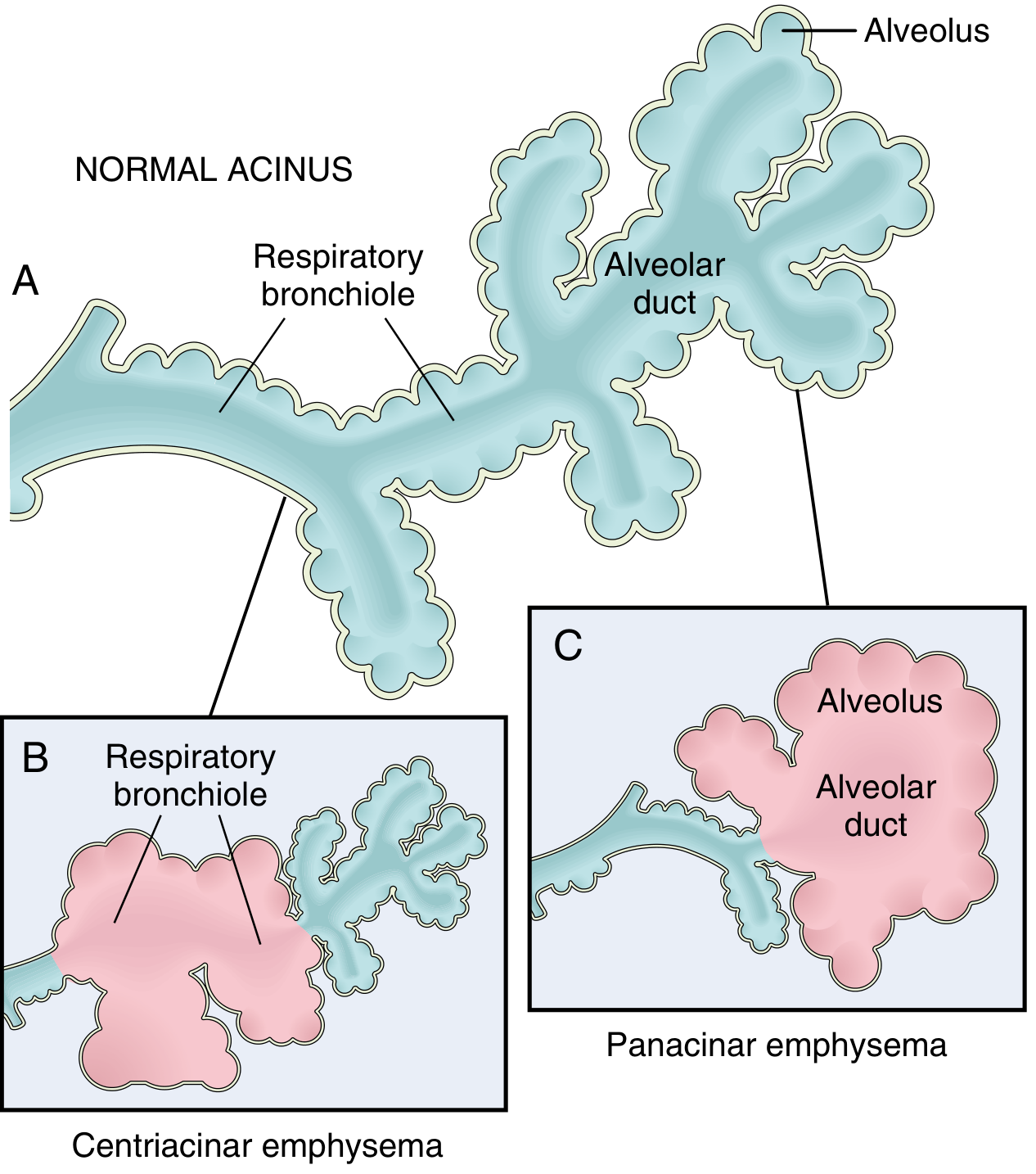
(A) Normal acinus. (B) Centriacinar emphysema: respiratory bronchioles dilated/destroyed (pink = affected; blue = spared distal alveoli). (C) Panacinar emphysema: entire acinus uniformly affected.
Gross pathology of emphysema (Robbins Fig. 15.7):
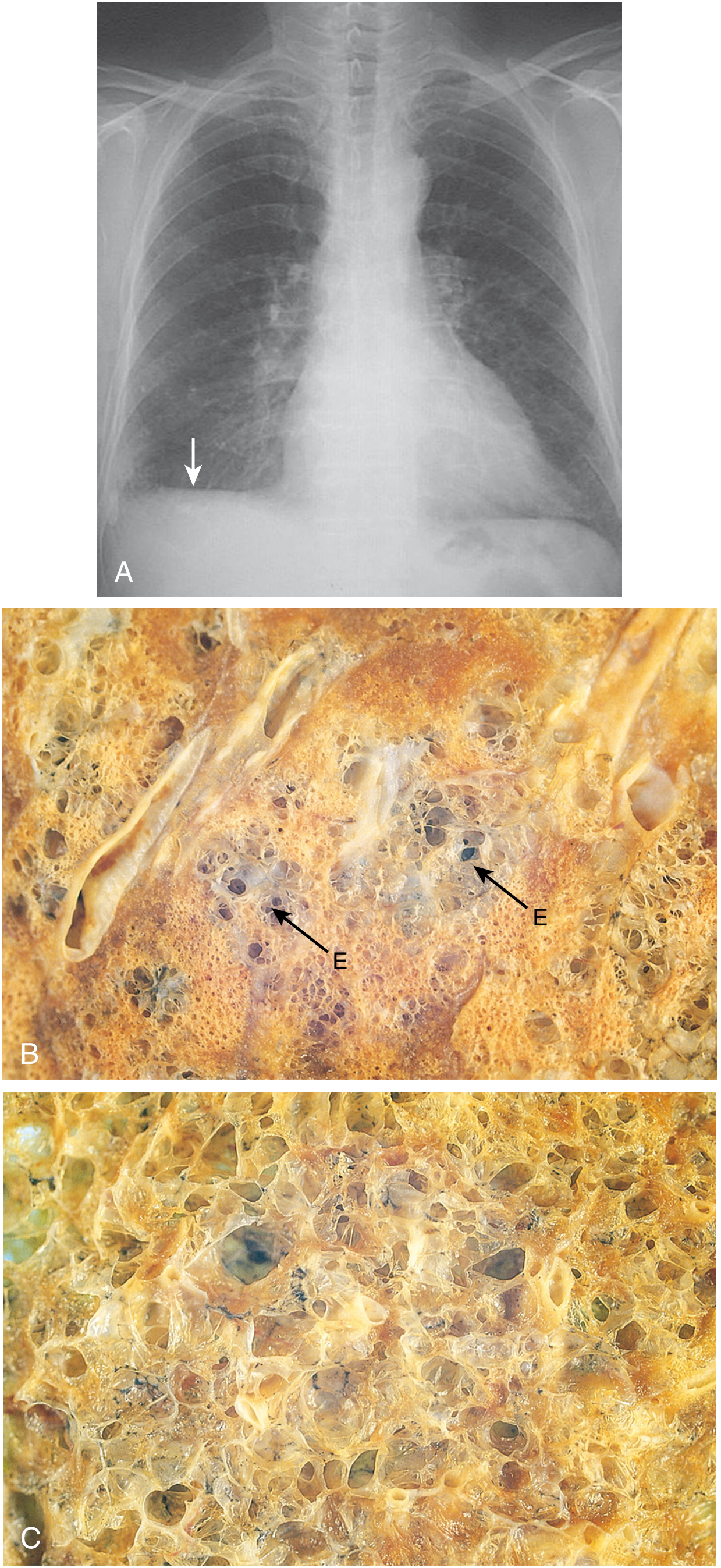
(A) CXR: flattened diaphragm (arrow). (B) Centriacinar emphysema: central areas show emphysematous spaces (E) surrounded by relatively preserved distal alveoli. (C) Panacinar emphysema: uniformly enlarged airspaces throughout the lobule.
Post-Streptococcal GN - Microscopy Image from Robbins:
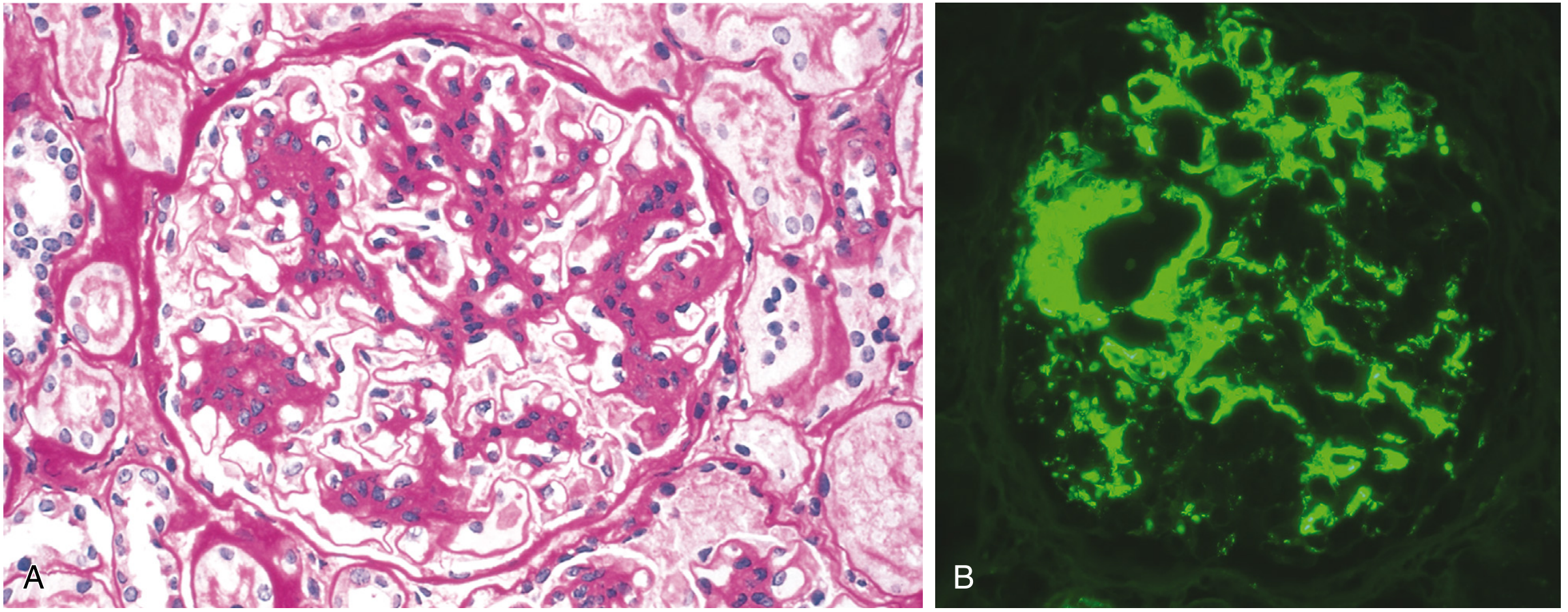
(A) H&E: mesangial proliferation and matrix increase in IgA nephropathy. (B) Immunofluorescence: characteristic mesangial IgA deposition (bright green granular deposits in mesangium) - pathognomonic for IgA nephropathy.
Glomerular Diseases (Chapter 20 - extended)
Pyelonephritis:
- Ascending route (most common): E. coli → colonizes urethra → bladder → kidney
- Predisposing: vesicoureteral reflux, obstruction, catheterization, female sex (short urethra)
- Hematogenous route: less common; septicemia, endocarditis; staphylococci, fungi
Acute Pyelonephritis - Morphology:
- Patchy suppurative inflammation; PMNs within tubular lumens and interstitium
- White cell casts (tubular leukocyte casts) - pathognomonic of pyelonephritis
- Abscesses in cortex and medulla
- Stain: H&E (suppurative inflammation, WBC casts); PAS (disrupted tubular BM)
Chronic Pyelonephritis:
- Coarse, irregular cortical scarring with underlying blunted, deformed calyx
- Microscopy: tubular atrophy, interstitial fibrosis; "thyroid-like" tubules (dilated tubules filled with eosinophilic casts resembling thyroid follicles) = characteristic
- Stain: H&E; Masson Trichrome for interstitial fibrosis
Acute Tubular Injury (ATI) / ATN:
(Full detail in previous notes)
- Key stain: PAS - loss of brush border (proximal tubule)
- Ischemic ATN: tubular rupture (tubulorrhexis); skip lesions; worst at corticomedullary junction
- Nephrotoxic ATN: uniform proximal tubule involvement; intact BM
CHAPTER 21 - GENITAL TRACT (Male and Female)
Male Genital Tract
Testicular tumors:
- Germ cell tumors (95%): Seminoma vs. Non-seminoma (NSGCT)
| Feature | Seminoma | NSGCT (Embryonal, Yolk sac, Choriocarcinoma, Teratoma) |
|---|---|---|
| Age | 30-40 | 20-30 |
| Gross | Homogeneous, cream-white | Variegated, hemorrhagic |
| Histology | Large cells with clear cytoplasm, central nuclei, "fried egg" appearance; lymphocytic stroma | Variable by type |
| Marker | PLAP, OCT3/4, D2-40 | AFP (yolk sac), hCG (choriocarcinoma), PLAP (some) |
| Radiosensitive | Yes | No |
- Seminoma stain: H&E (lymphocytic fibrous stroma, sheets of large clear cells); IHC: PLAP+, OCT3/4+, D2-40+, CD117+, AFP-, hCG-
- Yolk sac tumor (endodermal sinus tumor): Schiller-Duval bodies (glomeruloid structures); IHC AFP+
- Choriocarcinoma: syncytiotrophoblasts (multinucleated) + cytotrophoblasts; hCG+; most aggressive (hematogenous spread)
Prostate Carcinoma:
- Most common cancer in American males
- Peripheral zone (posterior lobe) - 70%; transition zone (anterior) - 20%
- Gleason grading (1-5 for predominant + secondary pattern = total score/Grade Group)
- Morphology: small, closely packed glands with prominent nucleoli; lacks basal cell layer; perineural invasion characteristic
- Stains: H&E (small glands, nucleoli); IHC: PSA+ (staining), p63-/HMWCK- (absent basal cells), AMACR (racemase, P504S) - positive in cancer, negative in benign; PIN4 cocktail (p63 + HMWCK + AMACR)
Female Genital Tract
Cervical Carcinoma:
- Most linked to HPV 16 and 18 (squamous carcinoma and adenocarcinoma)
- Progression: normal → HPV infection → LSIL (CIN1) → HSIL (CIN2/3) → invasive carcinoma
- Koilocytes (HPV cytopathic effect): squamous cells with perinuclear halo + wrinkled nucleus
- Stain: Pap smear (koilocytes, dysplastic cells); H&E (CIN grading, invasive carcinoma); p16 IHC (strong block positivity in HSIL/carcinoma - surrogate for high-risk HPV); Ki-67 (increased proliferation)
Endometrial Carcinoma:
| Type | Type I (Endometrioid) | Type II (Serous/Clear cell) |
|---|---|---|
| Background | Endometrial hyperplasia, obesity, estrogen | Atrophic endometrium |
| Genetics | PTEN, MSI, KRAS, CTNNB1 | TP53 (most common) |
| Grade | Low | High |
| Prognosis | Better | Poor |
- Stain: H&E (glandular proliferation, back-to-back glands, endometrioid carcinoma); IHC: ER+, PR+ (type I); TP53 mutant-type staining (type II); MMR proteins for Lynch screening
Ovarian Tumors:
-
Surface epithelial tumors (70%): serous (most common), mucinous, endometrioid, clear cell
-
Serous cystadenocarcinoma: Papillary architecture; psammoma bodies (concentric laminated calcifications) characteristic
-
Stain: H&E (papillary serous with psammoma bodies); IHC: PAX8+, WT-1+, CK7+, CA-125+; p53 aberrant staining (high-grade)
-
Germ cell tumors: Dermoid cyst (mature cystic teratoma) most common benign ovarian tumor; Dysgerminoma = malignant equivalent of seminoma
-
Sex cord-stromal tumors: Granulosa cell tumor (Call-Exner bodies = small follicle-like spaces with eosinophilic secretion in H&E; produces estrogen → precocious puberty or postmenopausal bleeding); Inhibin+ IHC
CHAPTER 22 - BREAST
Fibrocystic Changes
- Most common breast lesion in women 25-50 yr
- Non-proliferative: cysts, fibrosis, apocrine metaplasia - no increased cancer risk
- Proliferative without atypia: mild epithelial hyperplasia - slightly increased risk
- Atypical hyperplasia (ADH/ALH) - 4-5x increased cancer risk
Breast Carcinoma
Two main categories:
- In situ carcinomas: DCIS (ductal), LCIS (lobular) - no invasion
- Invasive carcinomas: Invasive ductal NOS (most common), Invasive lobular, Medullary, Mucinous, Tubular
Invasive Ductal Carcinoma (No Special Type, NST):
- Most common (75%); hard, gritty texture (desmoplastic stroma)
- Morphology: irregular nests/cords/glands; nuclear pleomorphism; mitotic figures
- Stain: H&E (infiltrative glands in desmoplastic stroma); IHC panels:
- ER/PR status (estrogen/progesterone receptors) - 70-80% positive
- HER2 (ERBB2) - amplified/overexpressed in 20%
- Ki-67 (proliferation index)
- Triple-negative (ER-/PR-/HER2-) - most aggressive; BRCA1 linked
Invasive Lobular Carcinoma:
- E-cadherin loss (CDH1 mutation) → cells don't form glands; grow as "single file/Indian file" pattern
- Dyscohesive cells in linear cords through stroma
- Stain: H&E (single file pattern); IHC: E-cadherin negative (diagnostic)
DCIS:
- Malignant cells confined to ductal system, no basement membrane breach
- Comedo type: central necrosis; calcifications ("malignant-type calcifications" on mammography)
- Stain: H&E (cells filling ducts, central necrosis + calcification); IHC: p63/HMWCK (absent myoepithelial layer distinguishes from benign lesions)
LCIS:
- Signet-ring cell morphology; dyscohesive; fills and distends acini
- IHC: E-cadherin negative
Molecular Subtypes of Breast Cancer (PAM50 gene signature):
| Subtype | ER/PR | HER2 | Ki-67 | Prognosis |
|---|---|---|---|---|
| Luminal A | + | - | Low | Best |
| Luminal B | + | ± | High | Intermediate |
| HER2-enriched | - | + | High | Poor |
| Triple-negative/Basal | - | - | High | Worst |
MASTER IMAGE GALLERY - KEY Robbins Pathology Microscopy
1. Emphysema - Types Diagram (Robbins Fig. 15.6)
- (A) Normal acinus: respiratory bronchiole + alveolar duct + alveoli all clearly defined
- (B) Centriacinar: respiratory bronchioles (pink/dilated) destroyed proximally; distal alveoli (blue) spared
- (C) Panacinar: entire unit uniformly enlarged from bronchiole to alveolus
2. Emphysema - CXR and Gross Pathology (Robbins Fig. 15.7)
- (A) CXR: flattened diaphragm (arrow) - classic sign of hyperinflation
- (B) Centriacinar (centrilobular): central emphysematous spaces (E = holes) surrounded by preserved alveolar parenchyma
- (C) Panacinar: uniformly enlarged, thin-walled airspaces throughout; no preserved islands of parenchyma
3. IgA Nephropathy - H&E and Immunofluorescence (Robbins Fig. 20.18)
- (A) H&E: mesangial cell proliferation + increased mesangial matrix; capillary lumens narrowed
- (B) Immunofluorescence (FITC-labeled anti-IgA): bright green granular deposits exclusively in mesangium - diagnostic of IgA nephropathy (Berger disease)
4. Cirrhosis - Masson Trichrome (Robbins Fig. 18.7)
- (A) Active cirrhosis: thick blue collagen bands (Masson Trichrome) surround and isolate rounded red hepatocyte regenerative nodules; complete fibrous septa; portal-to-central bridging
- (B) After 1 year abstinence: dramatic regression - only thin, incomplete blue fibers remain; nodular architecture partially restored; near-normal hepatic parenchyma visible
COMPREHENSIVE STAIN REFERENCE TABLE (Robbins Chapters 8-22)
| Stain | Color of Target | Diagnoses / Uses |
|---|---|---|
| H&E | Nuclei blue; cytoplasm pink | Universal baseline; all histopathology |
| Masson Trichrome | Collagen = blue/green; muscle = red | Cirrhosis, fibrosis staging, MI scar |
| PAS | Glycogen, GBM, fungi, mucins = magenta | GN (GBM), Barrett mucin, fungal infection |
| PASD | Same but glycogen digested; AAT globules stay | Alpha-1 antitrypsin deficiency |
| Jones Methenamine Silver (JMS) | GBM = black; deposits = holes/spikes | Membranous nephropathy, MPGN, FSGS |
| Congo Red (polarized) | Amyloid = apple-green birefringence | All amyloidosis (AL, AA, transthyretin) |
| Prussian Blue (Perls) | Iron = blue | Hemochromatosis, hemosiderosis |
| Rhodanine / Rubeanic acid | Copper = orange-red | Wilson disease |
| Orcein / Victoria Blue | HBsAg = brown; elastic fibers dark | Chronic HBV, AAT deficiency |
| Reticulin (Gordon-Sweet) | Reticulin fibers = black | Liver architecture, HCC (thick trabeculae), lymphoma |
| Alcian blue (pH 2.5) | Acid mucins = bright blue | Barrett goblet cells; intestinal metaplasia |
| Oil Red O / Sudan IV | Lipid/fat = red | Fatty liver (frozen section), foam cells |
| Modified Giemsa | H. pylori = dark blue curved rods | H. pylori gastritis |
| Warthin-Starry | Spirochetes, H. pylori, Legionella = black | H. pylori, Bartonella, syphilis |
| Ziehl-Neelsen (ZN) | Acid-fast bacteria (MTB, MAC) = red | TB, leprosy; negative in Crohn (vs. TB) |
| GMS / Grocott | Fungi = black | Aspergillus, Candida, Mucor, PCP |
| EVG / Verhoeff-van Gieson | Elastic fibers = black | Emphysema (elastic loss), aortic dissection, vasculitis |
| PTAH | Fibrin = blue; striations = blue | Fibrin in GN crescents, pericarditis |
| Von Kossa | Calcium phosphate = black | Metastatic calcification, fat necrosis in pancreatitis |
| Ubiquitin IHC | Mallory-Denk bodies, tau, TDP-43 = brown | Alcoholic hepatitis, neurodegeneration |
| p16 IHC | Strong diffuse block = brown | HSIL/cervical carcinoma (high-risk HPV surrogate) |
| AMACR (P504S) IHC | Cytoplasmic = brown | Prostate carcinoma (positive); absent in benign |
| E-cadherin IHC | Membrane = brown (positive) | Absent in lobular breast carcinoma; lost in diffuse gastric |
| WT-1 IHC | Nuclear = brown | Mesothelioma, serous ovarian Ca, nephroblastoma |
| Calretinin IHC | Nuclear + cytoplasmic = brown | Mesothelioma (vs. adenocarcinoma); granulosa cell tumor |
| CD30 / CD15 IHC | Membrane/Golgi = brown | Reed-Sternberg cells (Hodgkin lymphoma) |
| CD20 IHC | Membrane = brown | B-cell lymphomas (NHL) |
| GPC3 (glypican-3) IHC | Cytoplasmic = brown | HCC (positive); yolk sac tumor |
| HepPar-1 IHC | Cytoplasmic granular = brown | Hepatocellular differentiation (HCC) |
Source: Robbins, Cotran & Kumar Pathologic Basis of Disease (ISBN 9780443264528), Chapters 8 (Neoplasia), 14 (Blood Vessels), 15 (Heart/Congenital), 16 (Lymphoid/Myeloid neoplasms), 17 (GI tract), 18 (Liver), 19 (Pancreas), 20 (Kidney), 21 (Genital tract), 22 (Breast). Images directly from textbook figures as credited.
once check if you missed any details from the text robins
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
After a thorough re-check of all chapters against the Robbins text, here are all the missed/incomplete details, organized as supplementary notes to fill every gap:
ROBBINS - SUPPLEMENTARY & MISSED DETAILS
Complete Gap-Fill Across Chapters 8, 14–22
CHAPTER 8 - NEOPLASIA (Missed Details)
Tumor Immune Evasion Mechanisms (from text)
- Tumors downregulate MHC class I → avoid CD8+ T-cell recognition
- Expression of PD-L1 (CD274) → inhibits T-cell activation via PD-1 on T cells (basis of anti-PD-1/PD-L1 immunotherapy)
- Secretion of TGF-β and IL-10 → suppress immune effectors
- FOXP3+ regulatory T cells (Tregs) infiltrate tumor microenvironment → suppress anti-tumor immunity
Tumor Antigens (missed)
- Tumor-specific antigens (neoantigens): products of mutated genes unique to cancer cells → key targets for immune checkpoint therapy
- Tumor-associated antigens: shared between cancer and normal tissues (AFP in HCC, PSA in prostate, CEA in colorectal - all used as tumor markers)
Paraneoplastic Syndromes (missed completely)
Distant effects of tumors NOT caused by direct tumor mass or metastasis:
| Syndrome | Tumor | Mechanism |
|---|---|---|
| Hypercalcemia | Squamous cell carcinoma (lung, head/neck), breast, renal, myeloma | PTHrP secretion OR osteolysis |
| SIADH | Small cell lung carcinoma | Ectopic ADH |
| Cushing syndrome | Small cell lung carcinoma, carcinoid | Ectopic ACTH |
| Lambert-Eaton (myasthenic syndrome) | Small cell lung carcinoma | Abs against presynaptic Ca2+ channels |
| Trousseau syndrome (migratory thrombophlebitis) | Mucinous adenocarcinoma (pancreas, GI) | Procoagulant tumor-derived mucin |
| Polycythemia | Renal cell carcinoma, hemangioblastoma, HCC | Ectopic EPO |
| Carcinoid syndrome | Well-differentiated NETs with liver metastasis | Serotonin, bradykinin, histamine |
Grading vs. Staging (missed nuance)
- Grade = histologic degree of differentiation (WHO Grades 1-4 for most tumors; Gleason score for prostate)
- Stage = anatomic extent of spread; TNM system is universal; stage is the single most important prognostic determinant
CHAPTER 14 - BLOOD VESSELS (Missed Details)
Hypertension - Pathogenesis Expanded
Two categories of hypertension:
- Primary (essential) hypertension (~95%): polygenic; increased renin-angiotensin activity, sodium retention, adrenergic drive, endothelial dysfunction; associated with obesity, insulin resistance, family history
- Secondary hypertension (~5%): identifiable cause:
- Renovascular (renal artery stenosis) - most common secondary cause
- Renal parenchymal disease (CKD)
- Primary hyperaldosteronism (Conn syndrome) - hypokalemia + HTN
- Pheochromocytoma (catecholamines)
- Coarctation of aorta
- Hypothyroidism/hyperthyroidism
Hypertensive Vascular Morphology:
- Hyaline arteriolosclerosis - plasma protein insudation into arteriolar walls; PAS-positive homogeneous pink hyaline thickening of arteriolar walls; common in benign hypertension and diabetes
- Hyperplastic arteriolosclerosis - "onion-skin" concentric thickening of arteriolar walls; smooth muscle cell lamination; seen in malignant hypertension (diastolic BP >120 mm Hg)
- Fibrinoid necrosis - acute arteriolar damage in malignant hypertension
- Stains: H&E (hyaline pink); PAS (hyaline = magenta); Masson Trichrome (fibrinoid necrosis = red/fuchsinophilic)
Vascular Tumors (missed entirely)
Benign:
- Hemangioma: Most common vascular tumor; two types:
- Capillary hemangioma - lobular clusters of capillary-sized vessels lined by plump endothelium; in skin/subcutaneous tissue
- Cavernous hemangioma - large dilated thin-walled vascular channels (also most common benign liver tumor); can bleed; H&E: dilated channels with RBCs
- Pyogenic granuloma (lobular capillary hemangioma) - polypoid; skin/mucous membranes; associated with pregnancy; histology: lobular proliferation of capillaries in edematous stroma; H&E
- Lymphangioma - dilated lymphatic channels lined by endothelium; no RBCs in lumen; H&E + D2-40 IHC (marks lymphatic endothelium)
Malignant:
-
Angiosarcoma: Highly malignant tumor of endothelial cells
- Associated with: liver (vinyl chloride, arsenic, Thorotrast exposure), breast (post-radiation), lymphedema (Stewart-Treves syndrome)
- Morphology: irregular vascular channels lined by atypical pleomorphic endothelial cells; poorly differentiated areas appear as solid sheets
- Stains: H&E (vascular channels with malignant cells); IHC: CD31+ (most sensitive), CD34+, ERG+ (nuclear), FLI-1+ (nuclear); Factor VIII-related antigen
-
Kaposi Sarcoma (KS):
- Four clinical forms: classic (Mediterranean elderly men), African endemic, AIDS-related (most common), iatrogenic (immunosuppressed)
- All associated with HHV-8 (Kaposi sarcoma herpesvirus)
- Morphology: spindle-shaped cells forming vascular slits with RBC extravasation; hemosiderin deposition; "promontory sign" (vascular channels projecting into dilated spaces)
- Stains: H&E; IHC: HHV-8 latent nuclear antigen (LANA-1) - pathognomonic positive; CD31+, CD34+
CHAPTER 15 - HEART (Missed Details)
Infective Endocarditis (IE) - Detailed
Organisms by clinical setting:
| Setting | Most Common Organism |
|---|---|
| Previously abnormal valve | Streptococcus viridans (50-60%) |
| Overall most cases | Staphylococcus aureus |
| IV drug users | S. aureus (tricuspid valve) |
| Normal/abnormal valves | S. aureus |
| Prosthetic valve (early <2 months) | Staph epidermidis, S. aureus |
| Prosthetic valve (late >2 months) | Strep viridans |
| Colorectal cancer patient | Strep bovis/gallolyticus |
| HACEK organisms | H. influenzae, Actinobacillus, Cardiabacterium, Eikenella, Kingella |
Vegetations of IE:
- Large, irregular, destructive masses on valve cusps (can extend onto chordae)
- Composed of: thrombotic debris + organisms + fibrin
- Can cause: valve destruction/perforation, ring abscess, septic emboli
- Compare: Rheumatic = small warty; NBTE = small bland; Libman-Sacks = both sides of valve
Morphology:
- Acute IE: Bulky, destructive vegetations; underlying valve necrosis/perforation
- Subacute IE: Less destructive; may have healed areas
- Stain: H&E (fibrinous vegetation, organisms); Gram stain (bacteria); PAS (fungi); Warthin-Starry (some bacteria); Culture is essential
Complications:
- Regurgitation → acute heart failure
- Perforation of valves
- Septic emboli → brain, kidney, spleen infarcts
- Mycotic aneurysms (infected emboli weaken vessel walls)
- Glomerulonephritis (immune complex deposition from chronic bacteremia)
Non-Bacterial Thrombotic Endocarditis (NBTE) / Marantic Endocarditis
- Small (1-5 mm) sterile, bland thrombi on line of valve closure
- No inflammation, no valve destruction
- Seen in: cancer (especially mucinous adenocarcinoma, Trousseau), DIC, hyperestrogenic states, debilitated/cachectic patients
- Complication: systemic emboli to brain/kidney (despite being sterile)
Libman-Sacks Endocarditis (SLE)
- Small-to-medium sterile vegetations on both surfaces of leaflets (unlike other endocarditides)
- Due to immune complex deposition + fibrinoid necrosis
- Can cause valvular scarring similar to RHD (late)
- Associated with antiphospholipid antibody syndrome too
Carcinoid Heart Disease (missed in previous notes)
- Occurs in ~50% of patients with carcinoid syndrome
- Requires liver metastases (liver normally degrades serotonin, bradykinin, etc.)
- Mediators: serotonin (5-HT), kallikrein, bradykinin, histamine, tachykinins; urinary 5-HIAA correlates with severity
- Right heart exclusively affected (pulmonary vasculature degrades mediators before reaching left heart; left heart involved if R-to-L shunt or primary pulmonary carcinoid)
- Morphology: fibrous intimal plaques (glistening white) on tricuspid and pulmonary valves/endocardium; valve leaflets become thick, stiff, resembling pearly plaques
- Tricuspid regurgitation and pulmonary stenosis are classic findings
- Stain: H&E (fibrosis of valve leaflets/endocardium); Masson Trichrome (fibrous plaques = blue)
Hypertensive Heart Disease (missed details)
-
Left-sided HHD:
- Diagnostic criteria: (1) LV hypertrophy (concentric, without other pathology) + (2) clinical/pathologic evidence of HTN elsewhere
- Heart weight can exceed 500 g; LV wall thickness >2.0 cm
- Microscopy: myocyte transverse diameter increase; perivascular + interstitial fibrosis
- AHA threshold: >130/80 mmHg; European: >140/90 mmHg
- Outcomes: (1) normal longevity, (2) IHD due to potentiated atherosclerosis + increased O2 demand, (3) renal/cerebrovascular damage, (4) CHF or SCD
-
Right-sided HHD (Cor Pulmonale):
- Isolated hypertrophy/dilation of RV due to pulmonary hypertension from primary lung disease
- Causes: COPD, pulmonary fibrosis, recurrent pulmonary emboli, primary pulmonary hypertension
- Acute cor pulmonale: rapid RV dilation (e.g., massive pulmonary embolism)
- Chronic cor pulmonale: RV hypertrophy + dilation
Sudden Cardiac Death (SCD) - Expanded
- Defined as unexpected cardiac death within 1 hour of symptom onset (or within 24 hrs of last being well)
- 325,000 individuals/year in US; 4-5 million worldwide
- Mechanism: almost always lethal arrhythmia (ventricular fibrillation or asystole)
- #1 cause: CAD (usually chronic severe fixed stenoses; acute plaque rupture found in only 10-20%)
- 80-90% of successfully resuscitated patients have NO evidence of acute MI by enzymes or ECG
- In young patients: HCM, congenital coronary anomalies, myocarditis, channelopathies (LQTS, Brugada), mitral valve prolapse, pulmonary hypertension
- Treatment: ICD (implantable cardioverter-defibrillator)
CHAPTER 16 - WHITE CELLS & LYMPHOID NEOPLASMS (Missed Details)
Immune Cell Antigen Reference Table (from Robbins Table 13.5)
| CD Marker | Normal Distribution | Tumor Use |
|---|---|---|
| CD1 | Thymocytes, Langerhans cells | - |
| CD3 | All T cells | T-cell marker |
| CD4 | Helper T cells | T-cell subset |
| CD5 | T cells + small subset of B cells | CLL/SLL (B-cells aberrantly positive) |
| CD8 | Cytotoxic T cells | T-cell subset |
| CD10 | Pre-B cells + germinal center B cells | B-ALL, follicular lymphoma, DLBCL |
| CD19 | Pre-B to mature B cells (not plasma cells) | B-cell marker |
| CD20 | Pre-B cells to mature B (not plasma cells) | B-cell lymphomas; target of rituximab |
| CD21 | Mature B cells (EBV receptor) | - |
| CD23 | Activated B cells | CLL/SLL (positive) |
| CD11c | Granulocytes, monocytes; hairy cell leukemia | Hairy cell leukemia |
| CD15 | Granulocytes; RS cells | Hodgkin lymphoma |
| CD30 | Activated lymphocytes; RS cells | Hodgkin lymphoma; ALCL |
| CD33 | Myeloid progenitors | AML |
| CD34 | Hematopoietic progenitors | Stem cell marker |
| CD56 | NK cells + subset of T cells | NK/T-cell lymphoma; myeloma |
| CD117 | Mast cells, hematopoietic progenitors, GISTs | Seminoma, AML, GIST |
| CD138 | Plasma cells | Myeloma |
ALL (Acute Lymphoblastic Leukemia/Lymphoma) - Expanded
- Most common cancer of children (peak age 3 years for B-ALL)
- 85% B-ALL; 15% T-ALL (adolescent males, presents as mediastinal/thymic mass)
- Key cytogenetics:
- t(12;21) ETV6::RUNX1 - most common translocation in childhood B-ALL; favorable prognosis
- t(9;22) BCR::ABL1 (Philadelphia chromosome) - in 25% of adult ALL; adverse prognosis; treated with TKIs (imatinib, dasatinib)
- Hyperploidy (>50 chromosomes) - favorable prognosis in children
- Hypoploidy - adverse prognosis
- KMT2A (MLL) rearrangements - infant ALL; poor prognosis
- NOTCH1 mutations - most T-ALLs
- Morphology: lymphoblasts with scant cytoplasm, fine chromatin, inconspicuous nucleoli, numerous mitoses; TdT+ (terminal deoxynucleotidyl transferase) - pathognomonic nuclear marker of lymphoblasts
- Stains: H&E; PAS (lymphoblasts show block positivity in B-ALL); IHC: TdT+ (nuclear), CD10+, CD19+, PAX5+ (B-ALL); CD3+, TdT+ (T-ALL)
CLL/SLL (Chronic Lymphocytic Leukemia / Small Lymphocytic Lymphoma)
- Most common leukemia of adults (Western countries)
- Indolent tumor of mature B cells
- Morphology: monotonous small lymphocytes + "prolymphocytes" + "smudge cells" on peripheral blood smear (characteristic)
- Proliferation centers (pseudofollicles) in lymph nodes - pathognomonic
- IHC: CD5+, CD23+, CD20+ (weak), CD19+, BCL-2+; CD10-; cyclin D1- (negative distinguishes from Mantle cell)
- Complications: hypogammaglobulinemia → infections; autoimmune hemolytic anemia (warm AIHA); Richter transformation → DLBCL (very aggressive)
Mantle Cell Lymphoma (MCL) - Missed
- Moderately aggressive; t(11;14) cyclin D1/IgH → cyclin D1 overexpression
- Morphology: monotonous small-to-medium lymphocytes; mantle zone growth pattern; "blastoid" variant = more aggressive
- IHC: CD5+, CD19+, CD20+, CD23-, CD10-, cyclin D1+ (pathognomonic), SOX11+
Marginal Zone Lymphoma (MALT Lymphoma) - Missed
- Indolent; arises at chronic inflammatory sites
- Gastric MALT lymphoma: driven by H. pylori infection - eradication of H. pylori can cure early-stage disease
- Other sites: salivary gland (Sjögren), thyroid (Hashimoto), lung
- Morphology: lymphoepithelial lesions - neoplastic B cells infiltrating and destroying epithelial glands; monocytoid B cells; plasma cell differentiation
- IHC: CD20+, CD19+; CD5-, CD23-, CD10-, cyclin D1-
- Stain: H&E (lymphoepithelial lesions); Giemsa (H. pylori in associated gastritis)
Hairy Cell Leukemia - Missed
- Very indolent; involves spleen and bone marrow predominantly
- BRAF V600E mutation in virtually all cases (diagnostic and therapeutic target)
- Morphology: medium-sized lymphocytes with oval/folded nuclei and abundant pale cytoplasm with "hairy" cytoplasmic projections (best seen on peripheral blood smear by phase contrast)
- Spleen: characteristic red pulp infiltration (not white pulp) → massive splenomegaly
- Marrow: classic "fried egg" appearance with surrounding halos and reticulin fibrosis → "dry tap" on aspiration
- IHC: CD11c+, CD25+, CD103+, CD123+, TRAP (tartrate-resistant acid phosphatase)+, cyclin D1+ (weak); Annexin A1+ (highly specific)
- TRAP stain (enzyme cytochemistry): positive in hairy cells (not done routinely now; IHC preferred)
Hodgkin Lymphoma - Full Subtype Table (Robbins Table 13.8)
| Subtype | Frequency | RS Cell Pattern | IF, Stains | EBV | Prognosis |
|---|---|---|---|---|---|
| Nodular Sclerosis | 65-70% (most common) | Lacunar cells in collagen bands; nodular | CD30+, CD15+, PAX5+ (weak) | 25% | Very good |
| Mixed Cellularity | 20-25% | Classic RS cells; mixed inflammatory background | CD30+, CD15+, PAX5+ (weak) | 70% | Good |
| Lymphocyte-Rich | 5% | RS cells; abundant lymphocytes | CD30+, CD15+, PAX5+ (weak) | 40% | Excellent |
| Lymphocyte-Depleted | <1% | Abundant RS cells; few lymphocytes | CD30+, CD15+ | 90% | Poor |
| Nodular Lymphocyte Predominant HL | 5% | LP cells ("popcorn cells") instead of RS cells | CD20+, CD79a+, BCL-6+; CD30-, CD15- | Rare | Excellent |
Reed-Sternberg cell IHC: CD30+ (membranous/Golgi), CD15+ (membranous/Golgi), PAX5+ (weak nuclear), EBV-LMP1+ (when EBV-related), CD45- (negative - distinguishes from NHL)
Multiple Myeloma - Expanded Morphology
- Bones: vertebrae, ribs, skull, pelvis, femur, clavicle, scapula (descending frequency)
- "Punched-out" lytic lesions on plain X-ray (1-4 cm); soft, gelatinous red tumor masses in bone
- Marrow: >30% plasma cells; eccentric nucleus, "clock-face" or "cartwheel" chromatin, perinuclear clearing (Golgi)
- Cytologic variants from abnormal Ig accumulation:
- Flame cells - fiery red cytoplasm
- Mott cells - multiple grapelike cytoplasmic droplets
- Russell bodies - cytoplasmic globular Ig inclusions
- Dutcher bodies - intranuclear Ig inclusions
- Amyloid (AL type) deposition in 10% → nephrotic syndrome, restrictive cardiomyopathy
- Stains: H&E (plasma cells); IHC: CD138+, CD38+, MUM-1+; kappa or lambda restriction (ISH or IHC); Congo Red if amyloid present
- CRAB criteria: Hypercalcemia, Renal failure, Anemia, Bone lesions
CHAPTER 17 - GI TRACT (Missed Details)
GI Neuroendocrine Tumors (Carcinoid Tumors) - Full Detail
-
Now termed: well-differentiated neuroendocrine tumors (WD-NETs); "carcinoid" still used colloquially
-
Location: >40% small intestine; then tracheobronchial tree/lung; gastric NETs
-
Gastric NETs associations: autoimmune atrophic gastritis, MEN1, Zollinger-Ellison syndrome; PPI therapy → ECL cell hyperplasia (minimal cancer risk)
-
Morphology:
- Yellow/tan, very firm (desmoplastic reaction); submucosal/intramural masses
- Histology: islands, trabeculae, glands, sheets of uniform cells with scant pink granular cytoplasm and "salt and pepper" chromatin (dispersed nuclear chromatin)
- Stains: H&E (uniform cells, salt-and-pepper nuclei); IHC: Synaptophysin+ and Chromogranin A+ (neuroendocrine markers); Ki-67 (grading: G1 <3%, G2 3-20%, G3 >20%)
- Poorly differentiated NETs = neuroendocrine carcinoma (NEC): high Ki-67, TP53 and RB mutations; small cell or large cell type
-
Carcinoid Syndrome (occurs <10% of patients):
- Requires liver metastases (first-pass portal metabolism bypassed)
- Symptoms: cutaneous flushing, sweating, bronchospasm, diarrhea, right-sided valvular fibrosis
- Marker: urine 5-HIAA (5-hydroxyindoleacetic acid, serotonin metabolite)
- Treatment: octreotide (somatostatin analogue)
Appendix (missed entirely)
-
Acute Appendicitis:
- Most common abdominal surgical emergency
- Pathogenesis: obstruction of lumen (fecalith, hyperplastic lymphoid tissue, tumor) → increased intraluminal pressure → mucosal ischemia → bacterial invasion
- Morphology: neutrophilic transmural inflammation; mucosal ulceration; periappendiceal fat inflammation (periappendicitis)
- Complications: perforation → peritonitis, pelvic abscess, pylephlebitis
- Stain: H&E (neutrophilic inflammation)
-
Carcinoid (NET) of Appendix:
- Most common appendiceal tumor; usually at the tip; <2 cm rarely metastasize
- Typical "carcinoid" morphology
-
Mucinous tumors of Appendix:
- Spectrum: mucinous adenoma → low-grade appendiceal mucinous neoplasm (LAMN) → mucinous adenocarcinoma
- Rupture of LAMN → pseudomyxoma peritonei (gelatinous mucinous ascites; "jelly belly")
- Stain: H&E + PAS (mucin = magenta); IHC: CK20+, CDX2+, CEA+
Anal Canal (missed entirely)
-
Squamous cell carcinoma of anus:
- Associated with HPV 16, 18 (same as cervical carcinoma)
- Risk factors: anal intercourse, HIV, immunosuppression
- Preceded by anal intraepithelial neoplasia (AIN) - analogous to CIN
- Stain: H&E; p16 IHC (positive - HPV surrogate)
- Treatment: Nigro protocol (chemoradiation - cisplatin + 5-FU + radiation) avoids surgery
-
Anorectal junction: transitional epithelium (cloacogenic); tumors here = basaloid/cloacogenic carcinoma (variant of squamous cell carcinoma)
CHAPTER 18 - LIVER (Missed Details)
Hepatic Adenoma - Expanded Molecular Subtypes (missed)
Four subtypes from Robbins text:
- HNF1A-inactivated (30-35%): Loss of HNF1A; female predominant; associated with MODY3; minimal HCC risk; steatosis prominent (Oil Red O positive)
- Inflammatory (35-50%): Activating mutations in gp130 (IL-6 coreceptor) → JAK-STAT activation; obesity/metabolic syndrome; elevated CRP/serum amyloid A; 10% have beta-catenin mutations → higher HCC risk; IHC: SAA (serum amyloid A) and CRP positive
- Beta-catenin-activated (15%): CTNNB1 (exon 3) mutations → nuclear beta-catenin; high HCC risk; males predominant; anabolic steroid use; IHC: nuclear beta-catenin positive, glutamine synthetase (GS) diffusely positive; exon 7/8 mutations = low activation = low HCC risk
- Sonic hedgehog (SHH): GLI1 fusion genes; high propensity for hemorrhage; bleeding risk even when small
Cavernous Hemangioma of Liver
- Most common benign liver tumor
- <2 cm; subcapsular; incidental finding on imaging
- Morphology: dilated thin-walled vascular channels with RBCs; separated by fibrous stroma
- Stain: H&E (dilated channels lined by flat endothelial cells, no atypia); IHC: CD31+, CD34+ (endothelial)
- Never biopsied (risk of catastrophic hemorrhage)
Metabolic Liver Diseases (missed in previous session)
Hemochromatosis:
- Hereditary: HFE gene mutation (C282Y most common, especially in Northern Europeans) → excessive intestinal iron absorption
- Iron deposits in: liver (periportal hepatocytes first), pancreas (diabetes), heart (DCM), skin (bronze discoloration), joints, pituitary/gonads
- Stain: Prussian Blue (Perls) - iron = bright blue in hepatocytes; Grades 1-4 by zonal distribution
- Grading: Grade 1 (periportal only) → Grade 4 (all zones + fibrous septa + bile duct epithelium)
Wilson Disease:
- Autosomal recessive; ATP7B gene mutation → defective biliary copper excretion → copper accumulates in liver, brain (basal ganglia), eyes (Kayser-Fleischer rings)
- Stain: Rhodanine or Rubeanic acid - copper = orange-red granules in hepatocytes
- Orcein stain - copper-binding protein positive
- Also: liver biopsy copper quantification (>250 mcg/g dry weight = diagnostic)
- Histology: steatosis + Mallory-Denk bodies + glycogenated nuclei (in early disease)
Alpha-1 Antitrypsin (AAT) Deficiency:
- Autosomal recessive; SERPINA1 gene; Pi*ZZ phenotype most common (PiMZ = carrier)
- AAT accumulates in hepatocytes as misfolded protein (cannot be secreted)
- Stain: PASD (PAS with diastase) - PAS-positive, diastase-resistant globules in periportal hepatocytes - pathognomonic
- Orcein also highlights AAT globules
- IHC: anti-AAT antibody - cytoplasmic globular positivity
- Leads to neonatal hepatitis, childhood cirrhosis, and adult HCC even without cirrhosis
Metabolic Dysfunction-Associated Steatotic Liver Disease (MASLD / MAFLD / NASH):
- Spectrum: steatosis → steatohepatitis (MASH) → fibrosis → cirrhosis → HCC
- Morphology of MASH (H&E):
- Macrovesicular steatosis (>5% hepatocytes)
- Lobular inflammation (lymphocytes, neutrophils)
- Hepatocyte ballooning degeneration (key feature)
- Perisinusoidal/pericellular "chicken wire" fibrosis (zone 3; Masson Trichrome = blue)
- Mallory-Denk bodies (less prominent than alcoholic)
- NAS (NAFLD Activity Score): steatosis + lobular inflammation + ballooning = scored 0-8; ≥5 = MASH
- SAF score (Steatosis, Activity, Fibrosis) - alternative
- Key stains: H&E (ballooning, steatosis, lobular inflammation); Masson Trichrome (pericellular zone 3 fibrosis); PASD, Prussian Blue to exclude other causes
CHAPTER 19 - PANCREAS (Missed Details)
Pancreatic Cystic Neoplasms
Intraductal Papillary Mucinous Neoplasm (IPMN):
- Arises from main pancreatic duct or its branches; produces copious mucin
- Main duct IPMN: higher malignant potential (~70% have high-grade dysplasia or invasive carcinoma)
- Branch duct IPMN: lower risk; often "wait and watch" if <3 cm without worrisome features
- Morphology: papillary projections lined by mucinous columnar epithelium; mucin fills and dilates the duct
- Associated genetic changes: KRAS mutations most common; GNAS mutations (activating, codon 201) - seen in ~50%; characteristic of IPMN (not seen in MCN)
- Stain: H&E + PAS (mucin); IHC: MUC2, MUC5AC (intestinal and gastric foveolar subtypes)
Mucinous Cystic Neoplasm (MCN):
- Almost exclusively in women; body/tail of pancreas
- Pathognomonic: ovarian-type stroma beneath mucinous epithelium
- No communication with pancreatic ductal system (unlike IPMN)
- All MCNs should be resected due to malignant potential
- Stains: H&E (ovarian stroma + mucinous lining); IHC: ER+, PR+ in ovarian stroma (confirming diagnosis); MUC5AC+ in epithelium
Serous Cystadenoma:
- Almost always benign (serous cystadenocarcinoma extremely rare)
- Von Hippel-Lindau (VHL) gene mutations in sporadic cases; associated with VHL syndrome
- Morphology: innumerable small cysts lined by glycogen-rich cuboidal epithelium; central stellate scar with calcification; "honeycomb" appearance
- Stains: H&E (small cysts, cuboidal clear cells); PAS (glycogen-rich cells = magenta); IHC: MUC6+
Pancreatic Neuroendocrine Neoplasms (PanNENs) - Missed
From the book's Chapter 24 cross-reference, mentioned in block 11:
- Insulinoma - most common functioning PanNET; beta cells; hypoglycemia; Whipple triad (hypoglycemia symptoms + low blood glucose + relief with glucose); H&E: uniform cells; IHC: insulin+; usually benign (>90%)
- Gastrinoma - G-cell; causes Zollinger-Ellison syndrome (multiple refractory peptic ulcers, diarrhea); 25% associated with MEN1 (along with parathyroid and pituitary tumors); often malignant; IHC: gastrin+
- Glucagonoma - alpha cells; necrolytic migratory erythema + diabetes; IHC: glucagon+
- VIPoma - massive watery diarrhea (WDHA / Verner-Morrison syndrome); IHC: VIP+
- Non-functioning PanNETs - most common; present with mass effect; often large at diagnosis
PanNET Morphology:
- Amyloid deposition in insulinomas (islet amyloid polypeptide = IAPP); Congo Red positive
- Stains: H&E (salt-and-pepper chromatin, trabeculae/nests); IHC: Synaptophysin+, Chromogranin A+; Ki-67 for grading
CHAPTER 20 - KIDNEY (Missed Details)
Renal Cell Carcinoma (RCC) - Missed Entirely
Three main types:
| Type | % | Cell of origin | Genetics | Gross/Micro | Stains |
|---|---|---|---|---|---|
| Clear Cell RCC | 75% | Proximal tubule | VHL loss (3p25 deletion); also PBRM1, BAP1, SETD2 | Yellow, bright; clear cytoplasm (glycogen/lipid); compact nests/alveolar pattern; rich vasculature | H&E (clear cells); PAS (glycogen); CD10+, PAX8+, CA-IX+, CAIX+, CK7- (IHC) |
| Papillary RCC | 10-15% | Proximal tubule | Trisomies 7, 17; loss of Y; Type 2 papillary: CDKN2A loss, TFE3 fusions | Bilateral/multifocal; papillary architecture; foamy macrophages in papillary cores; hemosiderin; psammoma bodies | H&E; CK7+ (especially type 1), AMACR+, PAX8+, CD10+ |
| Chromophobe RCC | 5% | Intercalated cells of collecting duct | Multiple monosomies | Pale brown/tan; pale-staining cells with prominent cell membranes; "plant cell" appearance | H&E; Hale's colloidal iron (diffuse cytoplasmic staining); CK7+, CD117+, E-cadherin+; vimentin-; CD10- |
Renal Oncocytoma (benign):
- Mahogany brown tumor; central stellate scar
- Large cells with abundant eosinophilic granular cytoplasm (packed mitochondria)
- IHC: CD117+, CK7- (negative in nests); Hale's colloidal iron negative/limited
- EM: packed mitochondria (confirms diagnosis)
Wilms Tumor (Nephroblastoma):
- Most common renal tumor of children (peak age 3-4 years)
- Associated with: WT1 gene deletion (chromosome 11p13) - also in WAGR syndrome (Wilms + Aniridia + GU malformations + mental Retardation); WT2 - Beckwith-Wiedemann syndrome
- Classic triphasic histology: blastema (small blue cells) + epithelium (primitive tubules/glomeruli) + stroma (fibroblasts/muscle)
- Stains: H&E (triphasic pattern); IHC: WT1+ (nuclear); Blastemal cells CD56+
Diabetic Nephropathy (mentioned only briefly in text)
- Leading cause of chronic kidney failure in the US (40% of type 1 and type 2 diabetics develop advanced nephropathy)
- Pathology described in Chapter 24 (endocrine)
- Key morphologic features:
- Diffuse glomerulosclerosis - mesangial expansion + GBM thickening (most common)
- Nodular glomerulosclerosis (Kimmelstiel-Wilson nodules) - pathognomonic; nodular PAS-positive deposits in mesangium; associated with microaneurysms; "ball-and-plug" appearance
- "Capsular drop" lesion (homogeneous PAS-positive material between parietal epithelium and Bowman capsule)
- "Fibrin cap" lesion (homogeneous eosinophilic cap over glomerular capillaries)
- Arteriolar hyalinosis (both afferent AND efferent arterioles - efferent arteriolar hyalinosis = pathognomonic of diabetes)
- Stain: PAS (mesangial expansion, KW nodules = magenta); JMS (GBM thickening); H&E (all lesions)
Bladder Carcinoma (missed entirely)
- Urothelial (transitional cell) carcinoma (TCC) - ~90% of bladder tumors
- Risk factors: smoking (#1), aromatic amine exposure (aniline dyes), cyclophosphamide, Schistosoma haematobium (squamous cell type), chronic cystitis
- Two pathways:
- Low-grade papillary tumors: FGFR3 mutations → papillary low-grade, recurrent but rarely invasive
- High-grade/invasive carcinoma: TP53, RB mutations, CIS → flat high-grade → muscle-invasive
- CIS (carcinoma in situ) = flat, high-grade; appears red velvety; poor prognosis marker despite being non-invasive
- Morphology:
- Papillary: finger-like fronds with fibrovascular cores; lined by urothelium
- Flat CIS: full-thickness nuclear atypia; no papillary architecture; cells are dyscohesive
- Invasive: nests/cords/sheets infiltrating muscularis propria (detrusor muscle)
- Stains: H&E (grading, depth of invasion); IHC: CK7+, CK20+, GATA3+, p63+, uroplakin; p53 overexpression (high-grade); FISH for chromosome 3, 7, 9, 17 (UroVysion) - cytological detection in urine
SUPPLEMENTARY STAIN ADDITIONS (Missed from Chapter Content)
| Stain | Application | Color | Source |
|---|---|---|---|
| Hale's Colloidal Iron | Sialomucins in chromophobe RCC | Diffuse cytoplasmic blue-green | Renal Ch 20 |
| Tartrate-Resistant Acid Phosphatase (TRAP) | Hairy cell leukemia | Red (enzyme cytochemistry; now replaced by Annexin A1 IHC) | Ch 16 |
| Annexin A1 IHC | Hairy cell leukemia specific | Cytoplasmic brown | Ch 16 |
| LANA-1 (HHV-8) IHC | Kaposi sarcoma | Nuclear brown | Ch 14 |
| CA-IX (carbonic anhydrase IX) IHC | Clear cell RCC | Membrane brown | Ch 20 |
| GNAS mutation testing | IPMN (pancreas) | Molecular test | Ch 19 |
| Synaptophysin/Chromogranin IHC | Neuroendocrine tumors (any site) | Cytoplasmic brown | Ch 17,19 |
| 5-HIAA (urine) | Carcinoid syndrome | - (urine biochemical test) | Ch 17 |
| TdT IHC | B-ALL and T-ALL (lymphoblasts) | Nuclear brown | Ch 16 |
| MUC2, MUC5AC IHC | IPMN subtypes | Cytoplasmic brown | Ch 19 |
| ER/PR IHC in stroma | Mucinous cystic neoplasm of pancreas (ovarian stroma) | Nuclear brown | Ch 19 |
| Glutamine Synthetase (GS) IHC | Beta-catenin-activated hepatic adenoma | Diffuse cytoplasmic brown | Ch 18 |
| SAA/CRP IHC | Inflammatory hepatic adenoma | Cytoplasmic brown | Ch 18 |
SUMMARY OF ALL MAJOR GAPS NOW FILLED
| Chapter | What Was Missing |
|---|---|
| Ch 8 | Tumor immunity, PD-L1, paraneoplastic syndromes table, tumor markers |
| Ch 14 | Primary vs secondary HTN, vascular lesions (hyaline/hyperplastic arteriolosclerosis), all vascular tumors (hemangioma, angiosarcoma, Kaposi sarcoma with HHV-8/LANA-1) |
| Ch 15 (Heart) | Full IE (organisms, complications), NBTE/Libman-Sacks, Carcinoid heart disease, HHD (left and right/cor pulmonale), SCD full detail |
| Ch 16 | Full CD marker table, ALL cytogenetics (t(12;21), t(9;22), TdT), CLL/smudge cells/proliferation centers, Mantle cell lymphoma (cyclin D1), MALT/Marginal zone, Hairy cell (BRAF/Annexin A1/TRAP), HL subtype table with LP cells, Myeloma cytology (Flame/Mott/Russell/Dutcher bodies, CRAB) |
| Ch 17 | Full carcinoid/NET morphology + carcinoid syndrome (5-HIAA), Appendix (acute appendicitis, LAMN, pseudomyxoma peritonei), Anal canal (squamous Ca, HPV, AIN, Nigro protocol) |
| Ch 18 | Hepatic adenoma 4 molecular subtypes, Cavernous hemangioma of liver, Hemochromatosis (Prussian Blue grading), Wilson disease (Rhodanine), AAT deficiency (PASD globules), MASLD/NASH histology (NAS score, pericellular fibrosis) |
| Ch 19 | IPMN (KRAS+GNAS, main vs branch duct), MCN (ovarian stroma, ER/PR+), Serous cystadenoma (VHL, glycogen-rich cells, PAS), PanNETs (Insulinoma/Gastrinoma/Glucagonoma/VIPoma details) |
| Ch 20 | RCC (clear cell/VHL, papillary/trisomy 7&17, chromophobe/Hale's colloidal iron), Oncocytoma, Wilms tumor (triphasic/WT1), Diabetic nephropathy (Kimmelstiel-Wilson nodules/PAS), Bladder Ca (FGFR3 vs TP53, TCC pathways, GATA3 IHC) |
All additional content verified directly from Robbins, Cotran & Kumar Pathologic Basis of Disease (ISBN 9780443264528)
once check agin if anything missing then make the whole content in to 2 ppts without missing any information from strat to end
Reading File
Finding Sources
Reading File
Reading File
Reading File
Reading File
Running Command
Writing File
make a pdf combining whole matter
Reading File
Running Command
This is a shared conversation. Sign in to Orris to start your own chat.