Cachexia
Reading File
Finding Sources
Searching PubMed
cachexia pathophysiology treatment
Reading File
Reading File
Reading File
Reading File
Reading File
I now have rich content from multiple authoritative sources. Let me compile the full answer.
Cachexia
Definition
Cachexia is a complex metabolic syndrome characterized by loss of muscle mass (with or without loss of adipose tissue) that cannot be reversed by nutritional supplementation alone. It is driven by chronic systemic inflammation rather than simple starvation, and is a distinct entity from malnutrition - replacing calories does not restore lean body mass. - Yamada's Textbook of Gastroenterology, p. 1562
A common clinical definition requires involuntary weight loss >5-10% of premorbid body weight (corrected for fluid retention), with associated muscle wasting. More formal criteria also include fatigue, anorexia, elevated inflammatory markers (e.g., CRP), BMI <20, or sarcopenia on CT scan. - Harrison's Principles of Internal Medicine 22e, p. 3214
Epidemiology
- Affects up to 80-85% of patients with advanced cancer; responsible for roughly 30% of cancer deaths
- Most common in pancreatic, gastric, and lung cancers
- Up to 20% of all cancer deaths are strongly linked to cancer cachexia
- Also prevalent in: chronic heart failure (cardiac cachexia), CKD, COPD, HIV/AIDS (~30% despite antiretroviral therapy)
- Robbins, Cotran & Kumar Pathologic Basis of Disease, p. 310; Yamada's, p. 1563
Pathophysiology
Cytokine-Driven Catabolism
The central mechanism is a chronic inflammatory state driven by proinflammatory cytokines released by tumor cells and tumor-associated macrophages:
| Cytokine | Effect |
|---|---|
| TNF-α | Muscle breakdown, lipolysis, anorexia |
| IL-1 | Anorexia, fever, muscle wasting |
| IL-6 | Acute-phase response, muscle atrophy, hypoalbuminemia |
| IFN-γ | Catabolic state |
These cytokines activate the ubiquitin-proteasome pathway, leading to degradation of major skeletal muscle structural proteins such as myosin heavy chain. - Robbins Basic Pathology, p. 310; Goldman-Cecil Medicine, p. 1888
GDF-15 / GFRAL Axis
Growth differentiation factor-15 (GDF-15), secreted by tumor cells and macrophages, binds to the GFRAL receptor in the brainstem to suppress appetite. This is an emerging target - inhibitors of GDF-15 (e.g., ponsegromab) block cachexia in animal models and have shown benefit in early clinical trials. - Robbins Basic Pathology, p. 310; Harrison's 22e, p. 3218
Metabolic Alterations
- Increased glucose turnover and energy expenditure (note: despite what is often stated, total caloric consumption is not always elevated - the key is disordered energy utilization)
- Increased lipid mobilization and lipolysis
- Insulin resistance
- Activation of the JAK/STAT pathway
- Hypoalbuminemia and elevated acute-phase reactants (CRP, fibrinogen) - inversely related to albumin and prealbumin levels
- Malabsorption and impaired GI function (tumor infiltration, side effects of treatment)
Clinical Features
| Feature | Detail |
|---|---|
| Profound weight loss | >5-10% of body weight |
| Muscle wasting (sarcopenia) | Loss of skeletal muscle, including respiratory muscles |
| Loss of subcutaneous fat | Adipose wasting |
| Anorexia | Reduced appetite, often out of proportion to disease burden |
| Fatigue | Often severe |
| Anemia | Common accompaniment |
| Edema | Due to hypoalbuminemia |
| Death | From atrophy of diaphragm and respiratory muscles |
Cachexia is the most common paraneoplastic syndrome and a major independent risk factor for mortality. - Goldman-Cecil Medicine, p. 1888
Staging / Grading
A practical approach from Harrison's:
- Involuntary weight loss ≥5% of body weight
- Sarcopenia on CT imaging
- BMI <20
- Elevated CRP/inflammatory markers
- Anorexia and fatigue
Assessment
- History: Ask about appetite, weight change, food intake
- Physical exam: Document muscle wasting (temporalis, interossei, quadriceps), subcutaneous fat loss
- Labs: CRP, albumin, prealbumin, CBC (anemia), metabolic panel
- Imaging: CT can quantify skeletal muscle cross-sectional area at L3 vertebra (gold standard for sarcopenia)
Treatment
Treatment of the underlying disease is the most effective intervention; cachexia partially or fully reverses when the primary illness responds.
Symptomatic treatment is modest at best. Key options:
Pharmacological Agents
| Agent | Evidence | Notes |
|---|---|---|
| Olanzapine 2.5-5 mg/night | First-line (ASCO) | 60% of cancer patients gained ≥5% weight vs. 9% placebo in RCT |
| Megestrol acetate 400-800 mg/day | Moderate | Weight gain (adipose, not muscle), fluid retention, no survival benefit |
| Corticosteroids | Short-term only | Appetite improvement; toxicity limits long-term use |
| Anamorelin (ghrelin receptor agonist) | Positive trials | 100 mg orally once daily; increases lean body mass |
| Mirtazapine | Not recommended | Ineffective in large RCT |
| Ponsegromab (anti-GDF-15 monoclonal Ab) | Phase 2 (not FDA approved) | Improved appetite, weight, and physical activity |
- Harrison's 22e, p. 3218; Goldman-Cecil, p. 1888
Nutritional Approaches
- Dietary counseling: Associated with weight gain in some trials; can be reasonably offered but effect size is small
- Enteral feeding tubes and TPN: NOT recommended routinely; do not improve survival and may worsen comfort at end of life
- Nutritional intervention alone cannot reverse the catabolic state - Sleisenger & Fordtran's GI and Liver Disease, p. 3760-3761
Non-Pharmacological
- Exercise programs (resistance training) - emerging evidence for preserving muscle mass
- Multicomponent interventions combining nutrition + exercise + anti-inflammatory approaches are under study (see PMID 40012451 - systematic review of multicomponent interventions for cancer cachexia, 2025)
Key Distinctions
| Feature | Starvation | Cachexia |
|---|---|---|
| Primary driver | Caloric deficit | Systemic inflammation |
| Muscle loss | Preserved (fat lost first) | Prominent and early |
| Albumin | Near normal | Low |
| Response to nutrition | Yes, reverses | No, cannot be reversed |
| CRP | Normal | Elevated |
Summary
Cachexia is a life-threatening, inflammation-driven wasting syndrome that affects the majority of patients with advanced cancer and many with other chronic diseases. Its core mechanism involves cytokine-mediated (TNF-α, IL-1, IL-6) activation of the ubiquitin-proteasome system in skeletal muscle, with emerging roles for GDF-15/GFRAL signaling. Treatment remains challenging - olanzapine is the current best pharmacological option for appetite stimulation, anamorelin shows promise for lean mass, and nutritional support alone is insufficient. Reversing the underlying illness remains the most effective strategy.
Bleb in cell
Finding Sources
Finding Sources
Reading File
Reading File
Finding Sources
Reading File
Finding Sources
Loading Image
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Loading Image
I now have all the material needed for a thorough answer. Here is the full response:
Cell Blebs
What is a Bleb?
A cell bleb is a dynamic, bubble-like protrusion of the plasma membrane caused by local detachment of the lipid bilayer from the underlying actin cortex (the meshwork of actin filaments just beneath the membrane). The bleb expands outward due to intracellular hydrostatic pressure, then either retracts or is shed from the cell.
Blebs occur in two very different contexts:
- Reversible cell injury - an early warning sign of stress
- Apoptosis - a programmed, deliberate process leading to cell fragmentation
1. Blebs in Reversible Cell Injury
When a cell suffers sublethal stress (ischemia, toxins, ATP depletion), one of the earliest morphological changes is surface blebbing of the plasma membrane.
Mechanism
Ischemia causes ATP depletion, which leads to:
- Rho GTPase inactivation
- Activation and relocalization of ADF/cofilin (actin depolymerizing factor) to the apical membrane
- ADF/cofilin mediates depolymerization, severing, and capping of F-actin, destroying the microvillar actin core
- Loss of the actin cytoskeleton scaffold → membrane instability → blebbing
The blebs are membrane-bound extracellular vesicles that can be exfoliated into the tubular lumen or internalized and recycled. - Brenner and Rector's The Kidney, p. 1206
At this stage, blebbing is reversible - if the injury is removed, the actin cytoskeleton can be restored and the cell survives.
Histological Appearance (Kidney Tubules)
The classic teaching example is early ischemic injury in renal proximal tubules:
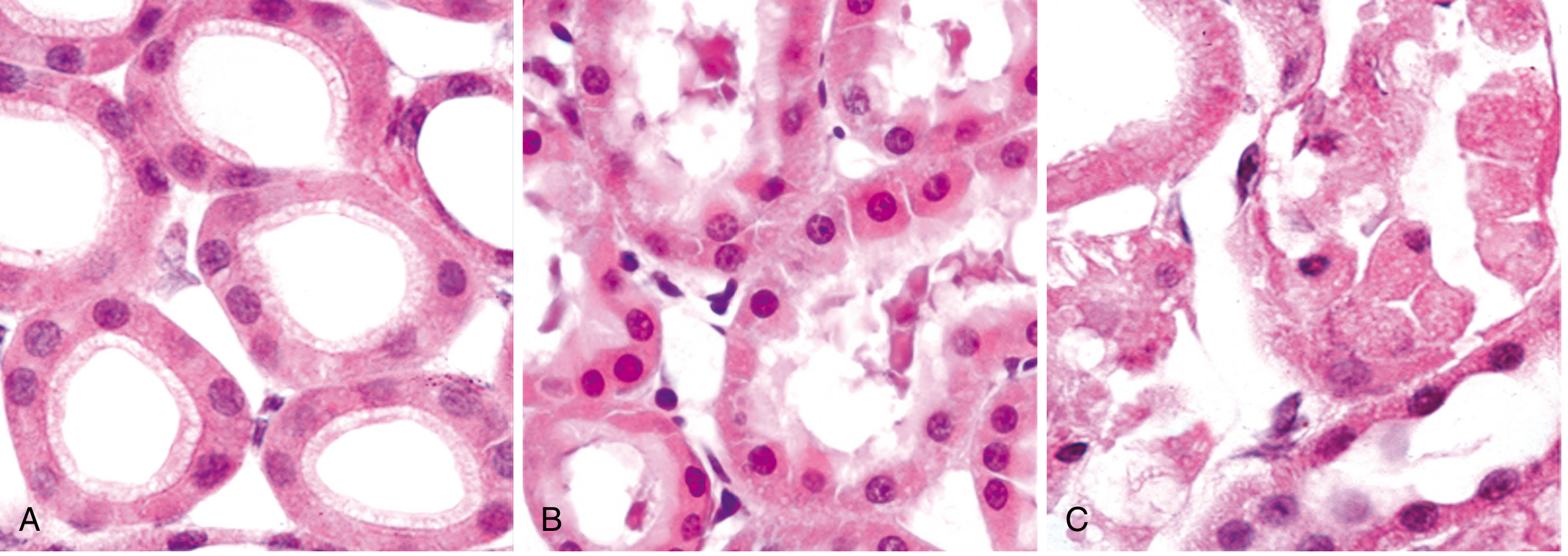
(A) Normal healthy tubular epithelial cells. (B) Early reversible ischemic injury - note surface blebs on the cell membrane, increased eosinophilia of cytoplasm, and cell swelling. (C) Irreversible necrosis - loss of nuclei, fragmentation, and leakage of contents. - Robbins, Cotran & Kumar Pathologic Basis of Disease, Fig. 2.8
2. Blebs in Apoptosis (Membrane Blebbing)
In apoptosis, membrane blebbing is a distinct, programmed morphological feature - and a key way apoptosis differs from necrosis.
Mechanism
Blebbing in apoptosis is driven by caspase activation:
- Caspases cleave cytoskeletal proteins (e.g., fodrin, gelsolin) that normally anchor the membrane to the actin cortex
- Phosphatidylserine (normally on the inner leaflet) translocates to the outer leaflet of the plasma membrane
- These membrane changes alter physical and chemical properties → blebbing without loss of membrane integrity (the membrane stays intact - no leakage, no inflammation)
- The blebs progressively enlarge and pinch off to form apoptotic bodies
- Histology: A Text and Atlas with Correlated Cell and Molecular Biology, p. 279
Apoptotic Bodies
-
The final step of apoptosis is cell breakage into apoptotic bodies
-
These are membrane-bounded extracellular vesicles containing organelles and nuclear fragments (condensed chromatin)
-
They are rapidly phagocytosed by macrophages and neighboring cells
-
Removal is so efficient that no inflammatory response is triggered
-
This is a fundamental distinction from necrosis, where membrane rupture releases contents and causes inflammation
-
Histology: A Text and Atlas, p. 280; Ganong's Review of Medical Physiology, p. 57
Electron Micrographs of Apoptotic Blebs and Bodies
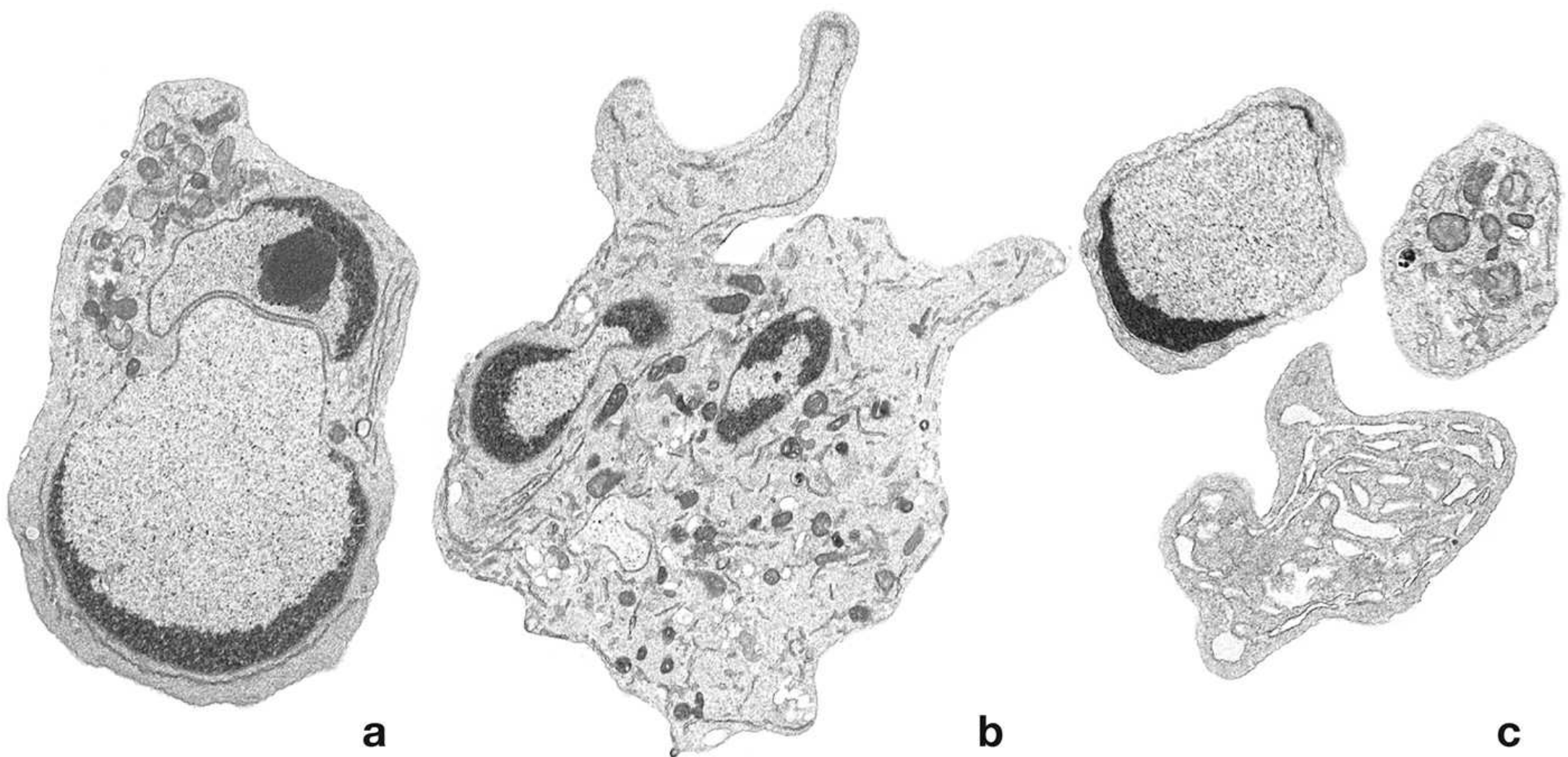
(a) Early apoptosis in a lymphocyte - nucleus already fragmented, condensed heterochromatin. (b) Further DNA fragmentation; heterochromatin buds outward, cytoplasm blebs. (c) Final apoptotic bodies containing nuclear fragments, organelles, and cytoplasm - ready for phagocytosis. - Histology: A Text and Atlas, Fig. 3.19
3. Necrosis vs. Apoptosis - Key Bleb Distinction
| Feature | Necrosis | Apoptosis |
|---|---|---|
| Plasma membrane blebbing | - (absent) | + (present) |
| Plasma membrane integrity | Disrupted (ruptures) | Intact |
| Cell size | Swells | Shrinks |
| Chromatin condensation | - | + |
| Inflammation | Yes (contents leak) | No (apoptotic bodies phagocytosed cleanly) |
| Caspase activation | - | + |
- Histology: A Text and Atlas, Table 3.2; Robbins Basic Pathology, Table 2.2
Summary
| Type | Context | Mechanism | Outcome |
|---|---|---|---|
| Injury blebs | Ischemia, ATP depletion, toxins | ADF/cofilin disrupts F-actin → membrane instability | Reversible if stress removed; irreversible → necrosis |
| Apoptotic blebs | Programmed cell death | Caspases cleave cytoskeletal anchors + phosphatidylserine externalization | Shed as apoptotic bodies → phagocytosis, no inflammation |
Cell blebs are therefore a morphological readout of membrane-cytoskeleton uncoupling - whether from pathological stress or controlled cell death signaling.

photo-1784294717200.jpg
Explain everything in easiest possible language in flowchart form
The image shows Mitochondrial Damage pathways from a pathology notes page. Let me explain everything in the simplest language possible as flowcharts.
🔋 Mitochondrial Damage — Simple Flowcharts
Think of mitochondria as the BATTERY of the cell. When the battery is damaged, 3 bad things happen:
PATH 1 — The Battery Dies (No Energy)
🔋 Mitochondria get damaged
↓
A "leak hole" (high-conductance channel)
opens in the mitochondrial membrane
= called the "Permeability Transition Pore"
↓
The membrane loses its seal
(like a battery leaking acid)
↓
Normal energy-making process
(oxidative phosphorylation) FAILS
↓
❌ No more ATP produced
= Cell runs out of energy
= Cell starts to die
Simple analogy: Your phone battery gets punctured → it can't hold charge → phone dies.
PATH 2 — The Battery Sparks (Toxic Waste)
🔋 Mitochondria get damaged
↓
Energy-making process goes WRONG
(abnormal oxidative phosphorylation)
↓
Produces toxic "sparks" called
Reactive Oxygen Species (ROS)
= basically chemical rust / free radicals
↓
⚠️ ROS damage DNA, proteins, membranes
= Harmful effects throughout the cell
Simple analogy: A faulty engine doesn't just stop — it also spews toxic exhaust fumes that damage everything around it.
PATH 3 — The Battery Sends a "Self-Destruct" Signal
🔋 Mitochondria get damaged
↓
Outer mitochondrial membrane
becomes MORE permeable (leaky)
↓
A protein called Cytochrome C
leaks out into the cell fluid (cytosol)
[normally it's locked between the
inner and outer membranes]
↓
☠️ Cytochrome C = "self-destruct signal"
↓
APOPTOSIS
= Cell commits controlled suicide
Simple analogy: The battery releases a chemical that tells the whole phone to shut itself down permanently and safely.
MASTER SUMMARY FLOWCHART
💥 MITOCHONDRIAL INJURY
↓
Opening of the Permeability
Transition Pore (leak hole)
↙ ↓ ↘
↙ ↓ ↘
❌ No ATP 💨 ROS made 🔓 Cytochrome C
(no energy) (toxic sparks) leaks out
↓ ↓ ↓
Cell can't Cell parts Apoptosis
function get damaged (cell suicide)
↓ ↓
Cell death Cell death
The 3 Outcomes — At a Glance
| Path | What happens | End result |
|---|---|---|
| Path 1 | No ATP made | Cell starves of energy → dies |
| Path 2 | ROS (toxic sparks) | DNA/protein/membrane damage |
| Path 3 | Cytochrome C leaks | Apoptosis (planned cell death) |
Key Terms — Plain English
| Term | Plain English |
|---|---|
| ATP | Cell's energy currency (like money for the cell) |
| Oxidative phosphorylation | The process that makes ATP inside mitochondria |
| Permeability transition pore | A leak hole that forms in the damaged mitochondrial membrane |
| ROS (Reactive Oxygen Species) | Toxic chemical sparks that damage cell components |
| Cytochrome C | A protein that, when released, triggers cell suicide |
| Apoptosis | Controlled, clean cell death (no inflammation) |
This is a shared conversation. Sign in to Orris to start your own chat.